

Replication fork stability confers chemoresistance in BRCA-deficient cells
- PMID: 27443740
- PMCID: PMC4959813
- DOI: 10.1038/nature18325
Replication fork stability confers chemoresistance in BRCA-deficient cells
Erratum in
-
Erratum: Replication fork stability confers chemoresistance in BRCA-deficient cells.Nature. 2016 Nov 17;539(7629):456. doi: 10.1038/nature19826. Epub 2016 Sep 28. Nature. 2016. PMID: 27680696 No abstract available.
Abstract
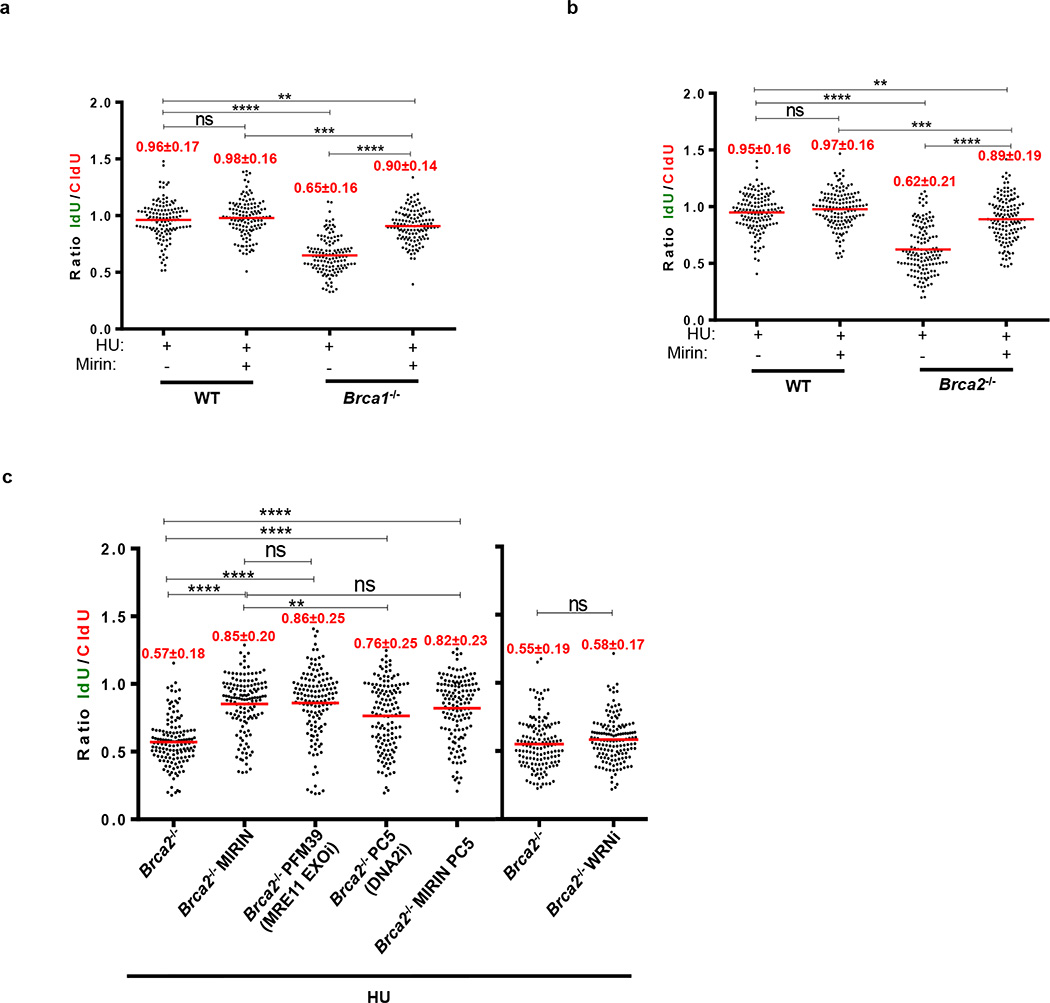
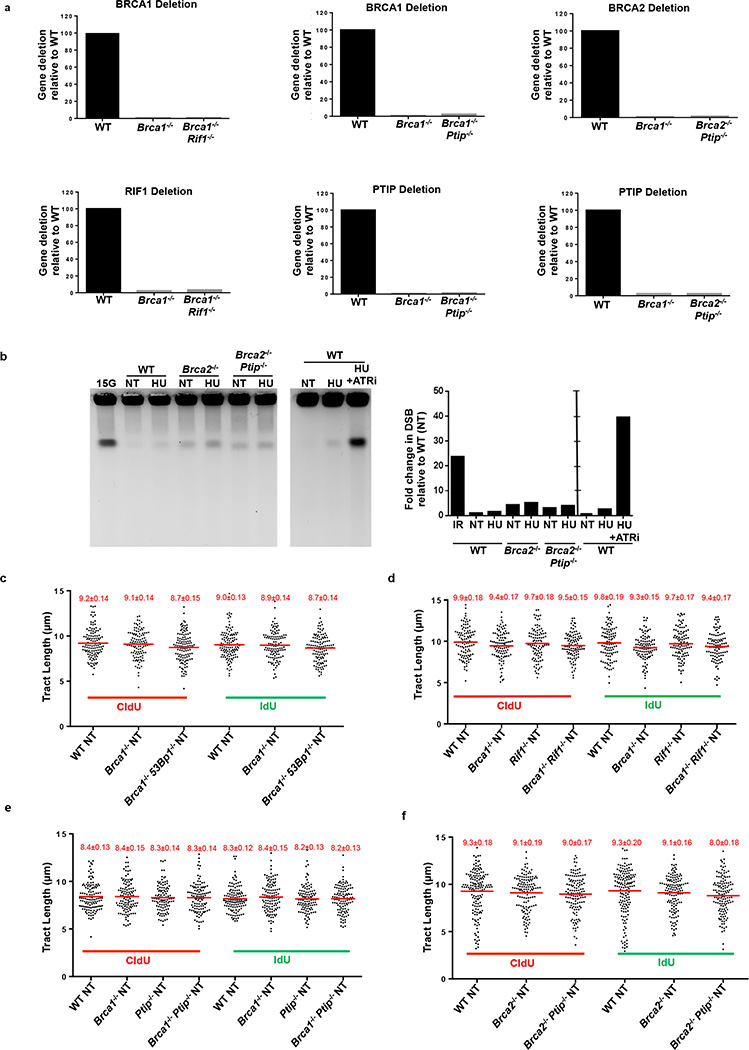
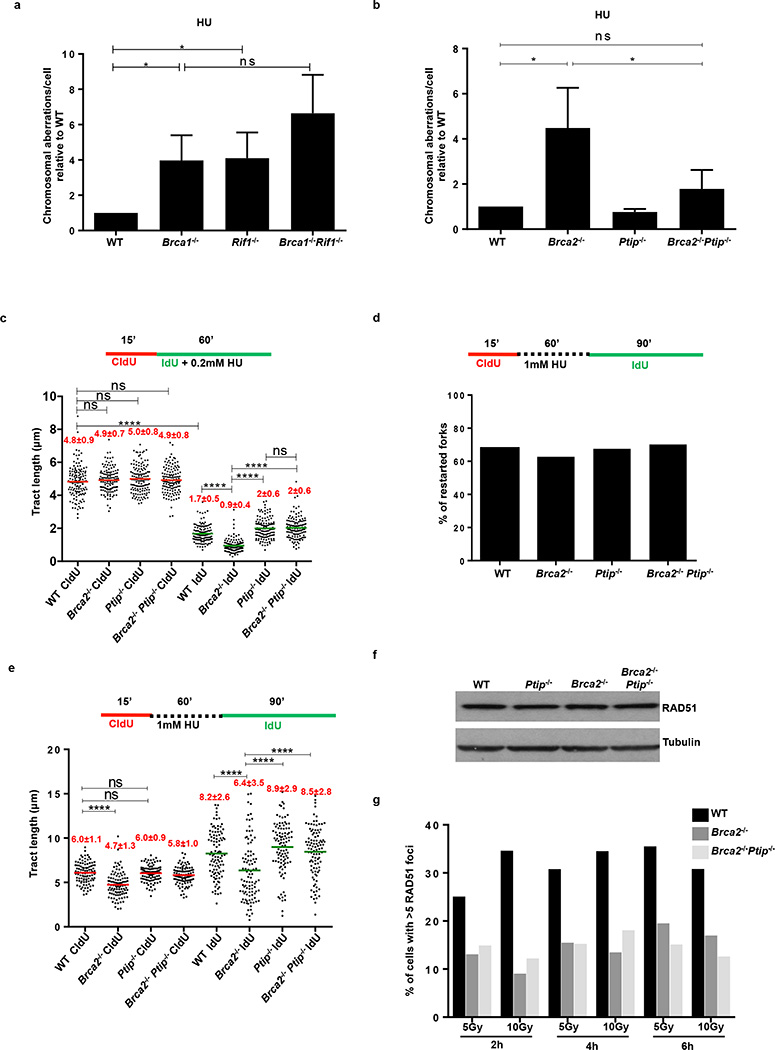
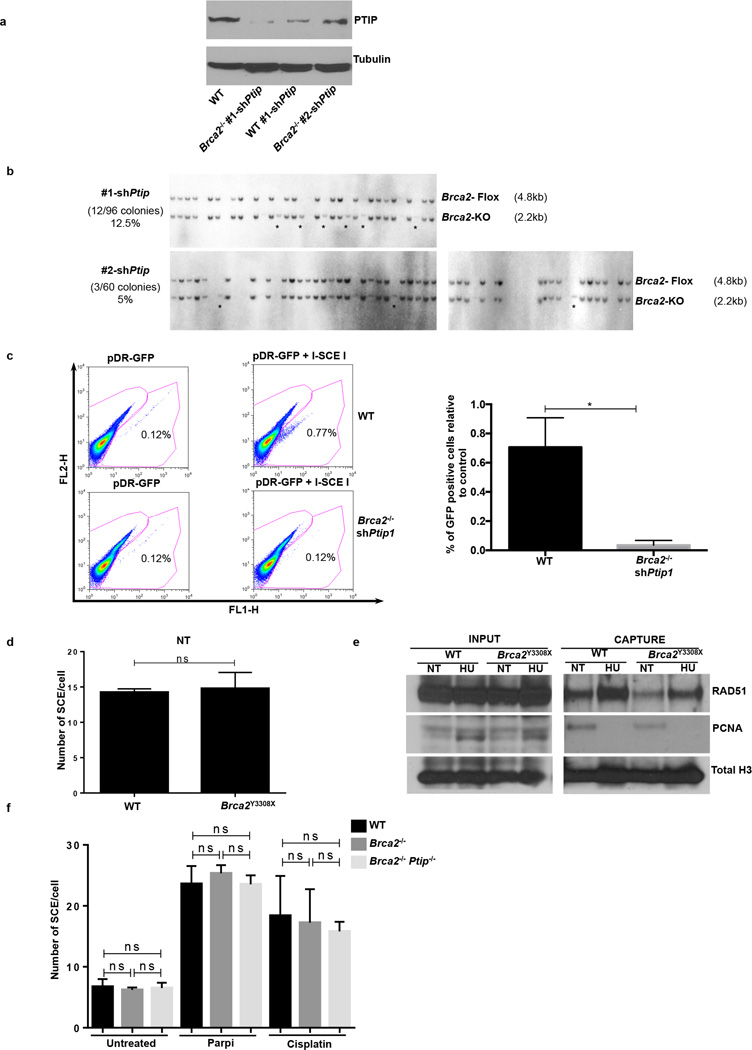
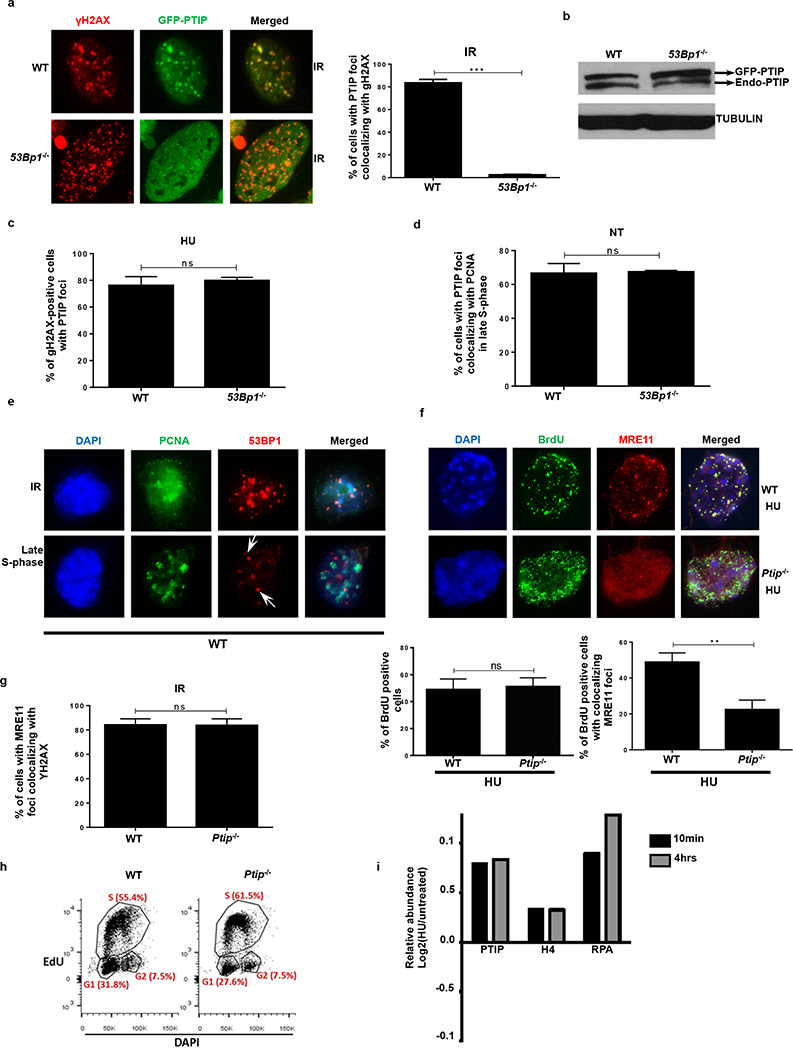
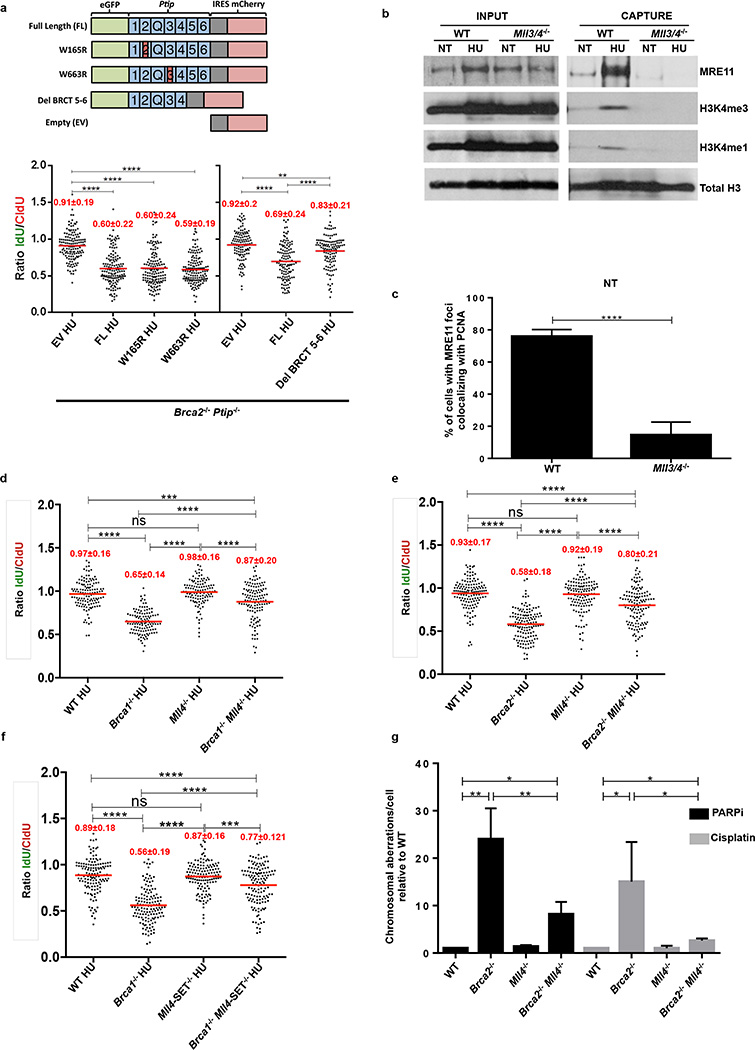
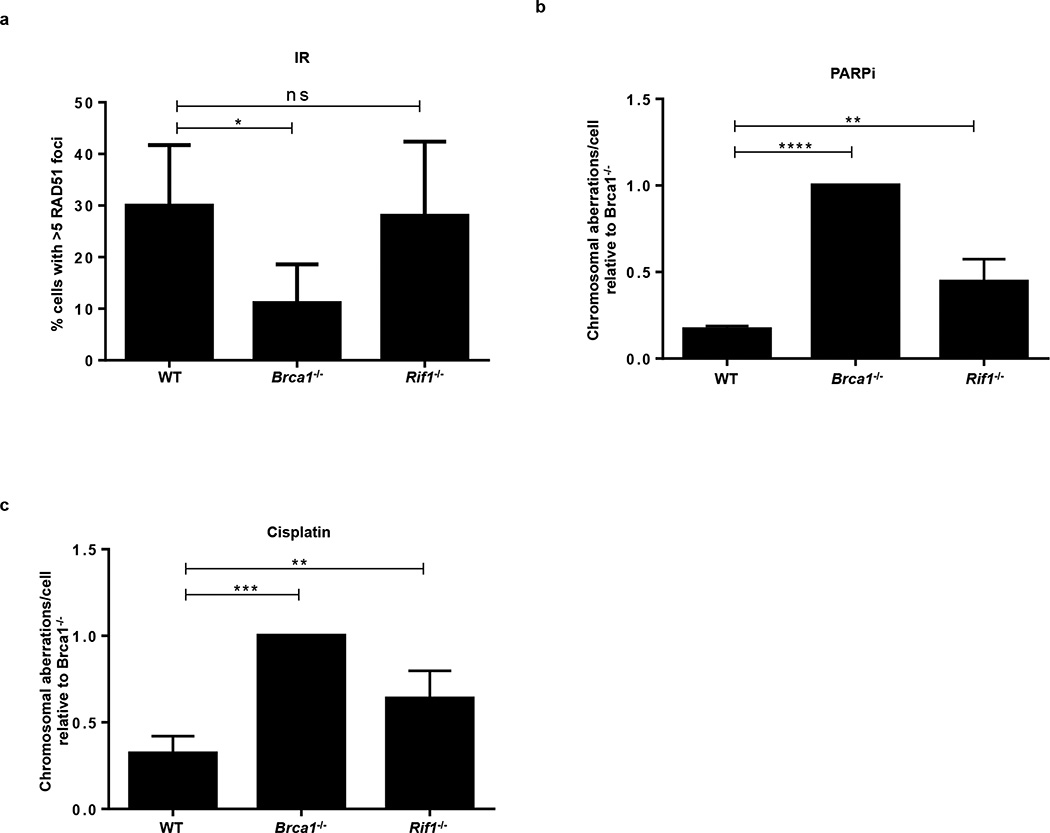
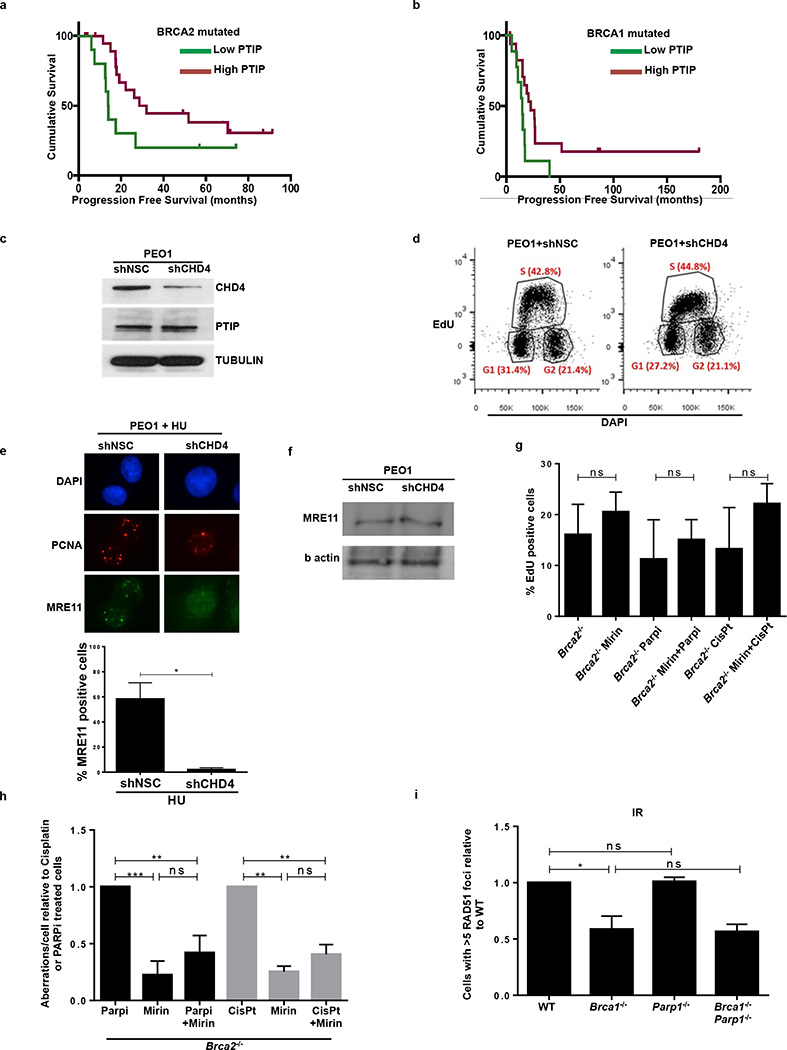
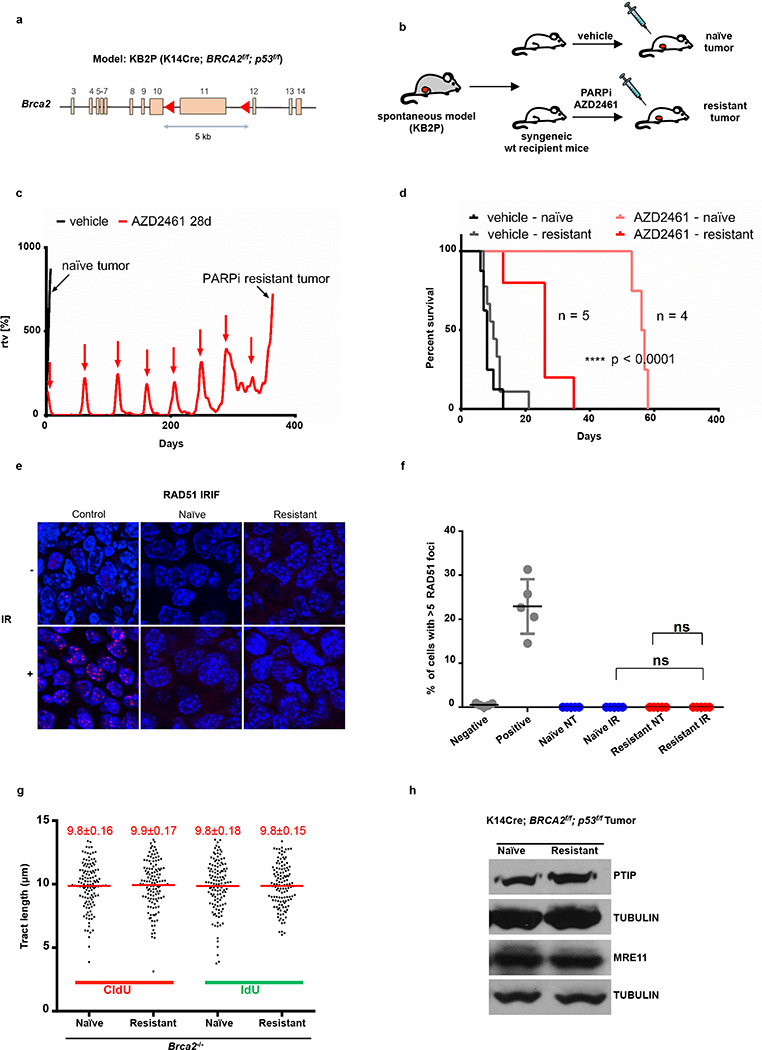
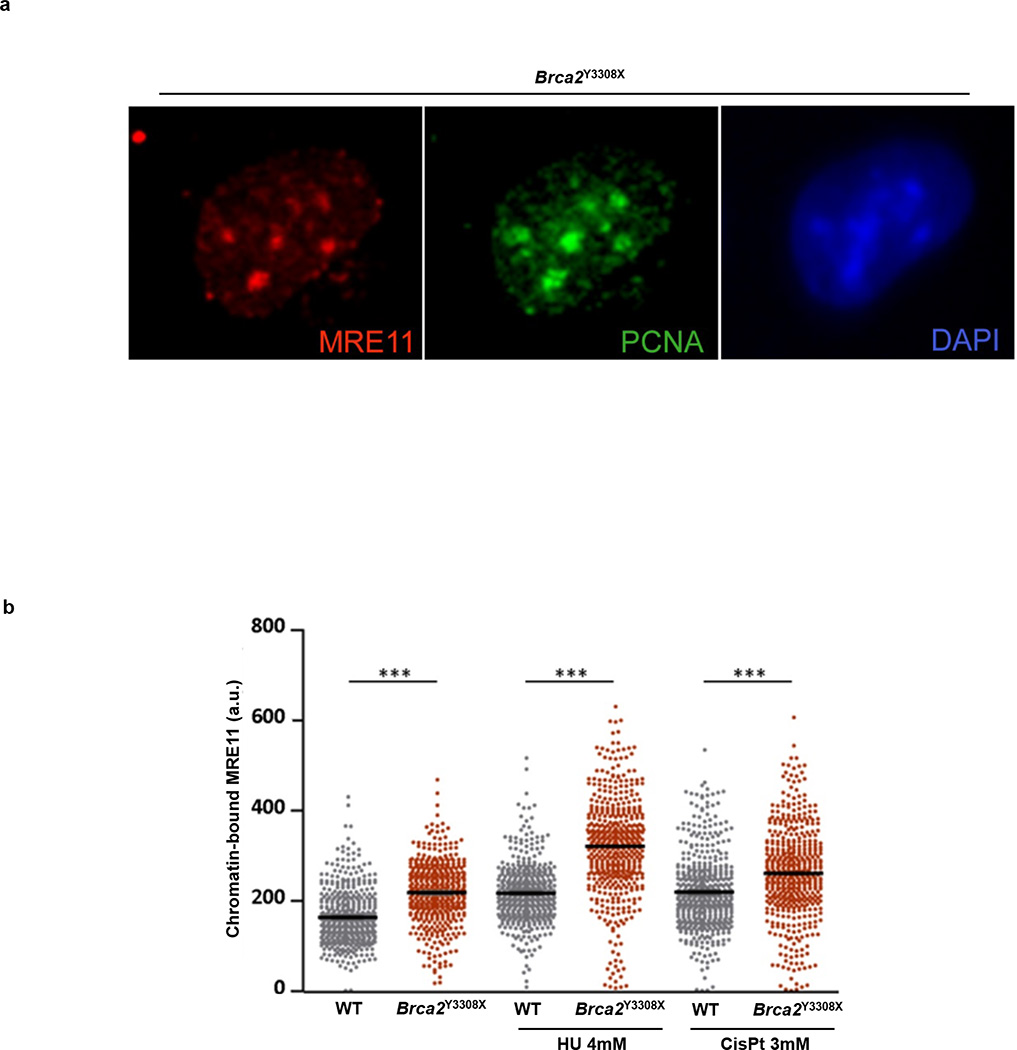
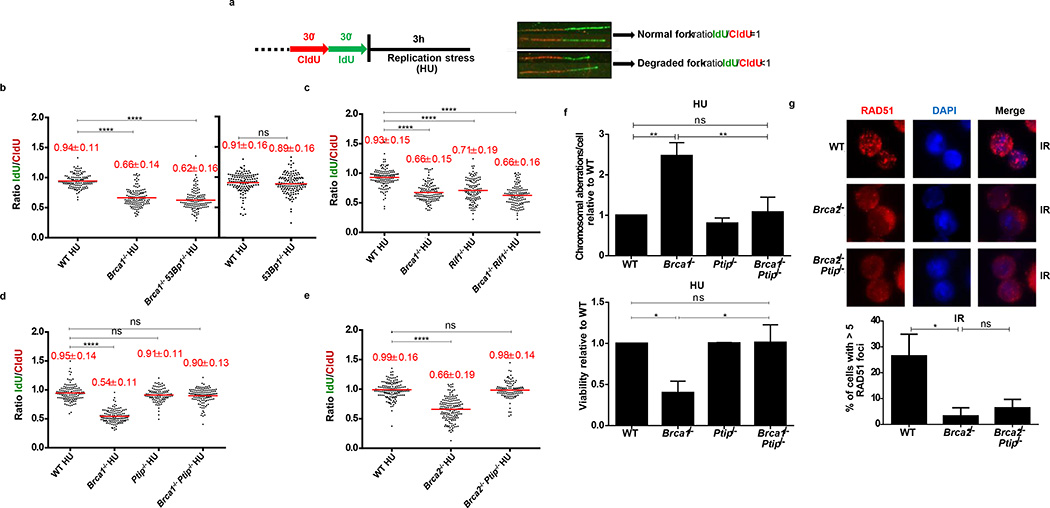
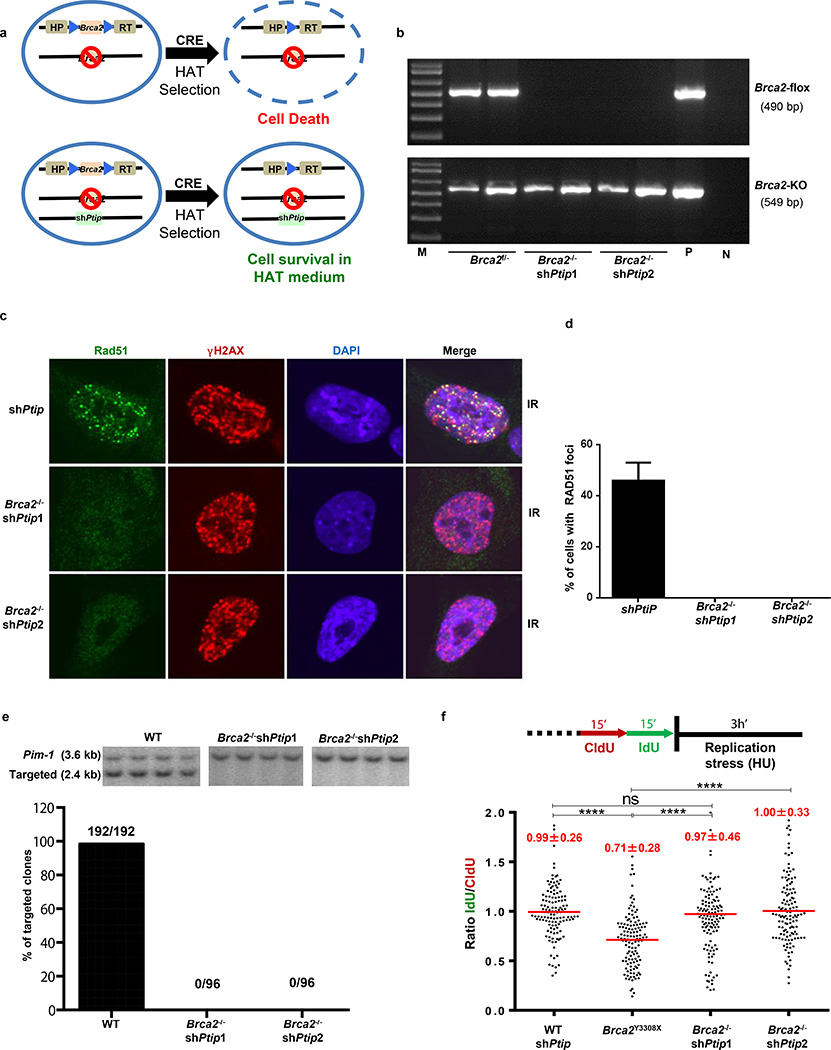
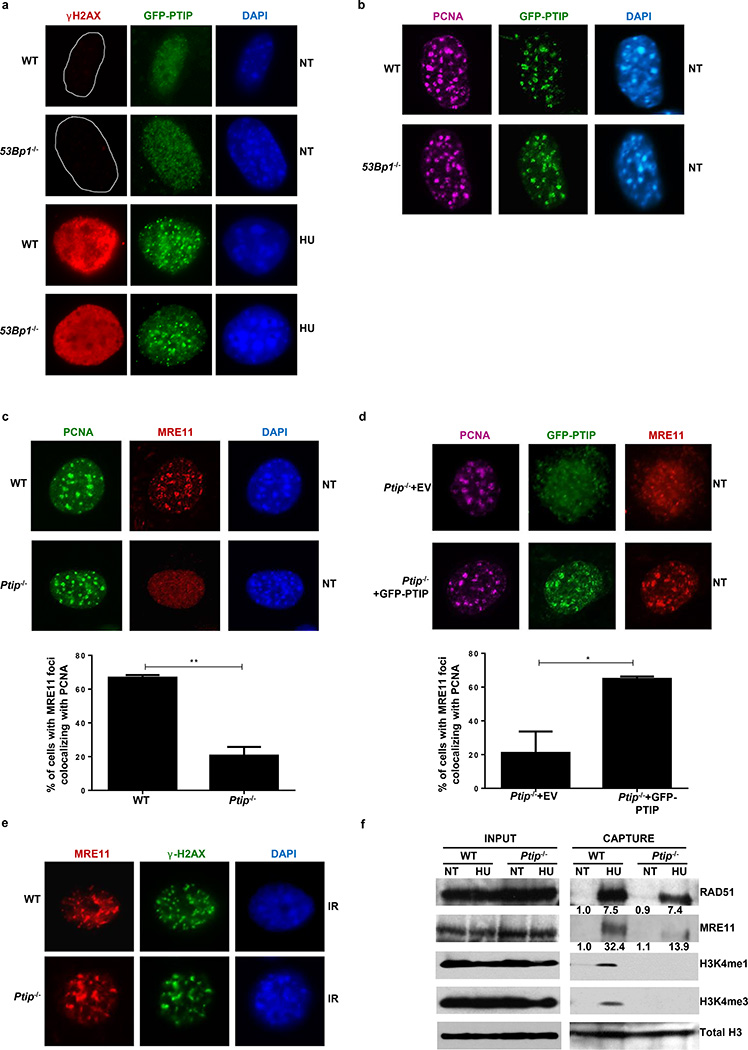
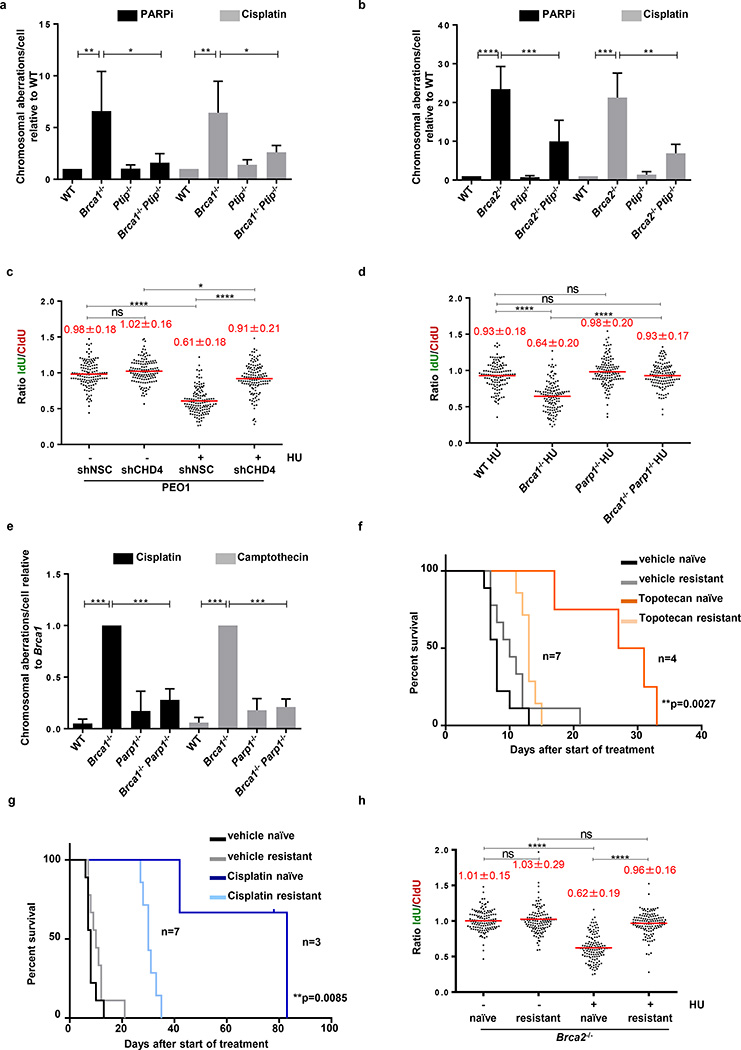
Cells deficient in the Brca1 and Brca2 genes have reduced capacity to repair DNA double-strand breaks by homologous recombination and consequently are hypersensitive to DNA-damaging agents, including cisplatin and poly(ADP-ribose) polymerase (PARP) inhibitors. Here we show that loss of the MLL3/4 complex protein, PTIP, protects Brca1/2-deficient cells from DNA damage and rescues the lethality of Brca2-deficient embryonic stem cells. However, PTIP deficiency does not restore homologous recombination activity at double-strand breaks. Instead, its absence inhibits the recruitment of the MRE11 nuclease to stalled replication forks, which in turn protects nascent DNA strands from extensive degradation. More generally, acquisition of PARP inhibitors and cisplatin resistance is associated with replication fork protection in Brca2-deficient tumour cells that do not develop Brca2 reversion mutations. Disruption of multiple proteins, including PARP1 and CHD4, leads to the same end point of replication fork protection, highlighting the complexities by which tumour cells evade chemotherapeutic interventions and acquire drug resistance.
Figures
References
-
- Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nature medicine. 2013;19:1381–1388. - PubMed
-
- Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer research. 2012;72:2814–2821. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
