

Activation of β-Glucocerebrosidase Reduces Pathological α-Synuclein and Restores Lysosomal Function in Parkinson's Patient Midbrain Neurons
- PMID: 27445146
- PMCID: PMC4951575
- DOI: 10.1523/JNEUROSCI.0628-16.2016
Activation of β-Glucocerebrosidase Reduces Pathological α-Synuclein and Restores Lysosomal Function in Parkinson's Patient Midbrain Neurons
Abstract
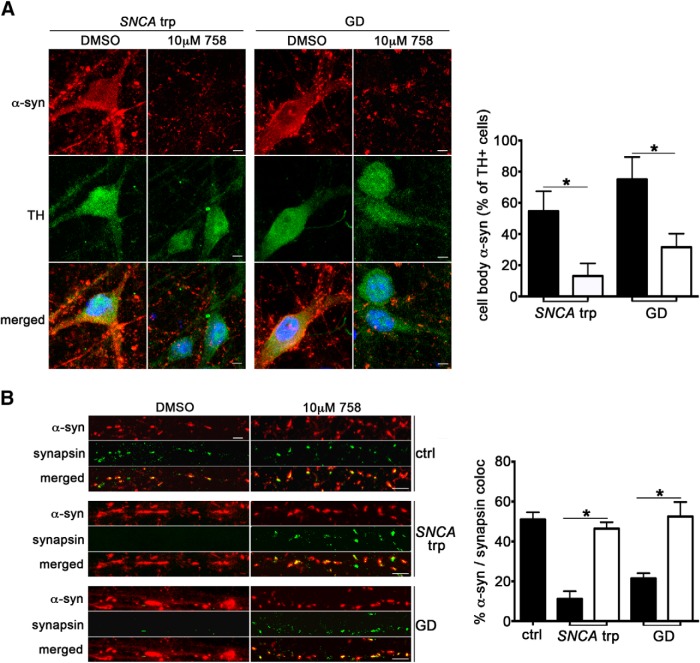
Parkinson's disease (PD) is characterized by the accumulation of α-synuclein (α-syn) within Lewy body inclusions in the nervous system. There are currently no disease-modifying therapies capable of reducing α-syn inclusions in PD. Recent data has indicated that loss-of-function mutations in the GBA1 gene that encodes lysosomal β-glucocerebrosidase (GCase) represent an important risk factor for PD, and can lead to α-syn accumulation. Here we use a small-molecule modulator of GCase to determine whether GCase activation within lysosomes can reduce α-syn levels and ameliorate downstream toxicity. Using induced pluripotent stem cell (iPSC)-derived human midbrain dopamine (DA) neurons from synucleinopathy patients with different PD-linked mutations, we find that a non-inhibitory small molecule modulator of GCase specifically enhanced activity within lysosomal compartments. This resulted in reduction of GCase substrates and clearance of pathological α-syn, regardless of the disease causing mutations. Importantly, the reduction of α-syn was sufficient to reverse downstream cellular pathologies induced by α-syn, including perturbations in hydrolase maturation and lysosomal dysfunction. These results indicate that enhancement of a single lysosomal hydrolase, GCase, can effectively reduce α-syn and provide therapeutic benefit in human midbrain neurons. This suggests that GCase activators may prove beneficial as treatments for PD and related synucleinopathies.
Significance statement: The presence of Lewy body inclusions comprised of fibrillar α-syn within affected regions of PD brain has been firmly documented, however no treatments exist that are capable of clearing Lewy bodies. Here, we used a mechanistic-based approach to examine the effect of GCase activation on α-syn clearance in human midbrain DA models that naturally accumulate α-syn through genetic mutations. Small molecule-mediated activation of GCase was effective at reducing α-syn inclusions in neurons, as well as associated downstream toxicity, demonstrating a therapeutic effect. Our work provides an example of how human iPSC-derived midbrain models could be used for testing potential treatments for neurodegenerative disorders, and identifies GCase as a critical therapeutic convergence point for a wide range of synucleinopathies.
Keywords: Parkinson's disease; glucocerebrosidase; induced pluripotent stem cells; lysosomes; synucleinopathy; α-synuclein.
Copyright © 2016 the authors 0270-6474/16/367694-14$15.00/0.
Figures







Similar articles
-
Glucocerebrosidase gene therapy prevents α-synucleinopathy of midbrain dopamine neurons.Neurobiol Dis. 2015 Oct;82:495-503. doi: 10.1016/j.nbd.2015.09.009. Epub 2015 Sep 25. Neurobiol Dis. 2015. PMID: 26392287
-
Increased Lysosomal Exocytosis Induced by Lysosomal Ca2+ Channel Agonists Protects Human Dopaminergic Neurons from α-Synuclein Toxicity.J Neurosci. 2019 Jul 17;39(29):5760-5772. doi: 10.1523/JNEUROSCI.3085-18.2019. Epub 2019 May 16. J Neurosci. 2019. PMID: 31097622 Free PMC article.
-
A novel glucosylceramide synthase inhibitor attenuates alpha synuclein pathology and lysosomal dysfunction in preclinical models of synucleinopathy.Neurobiol Dis. 2021 Nov;159:105507. doi: 10.1016/j.nbd.2021.105507. Epub 2021 Sep 9. Neurobiol Dis. 2021. PMID: 34509608
-
Parkinson's disease: acid-glucocerebrosidase activity and alpha-synuclein clearance.J Neurochem. 2016 Oct;139 Suppl 1:198-215. doi: 10.1111/jnc.13517. Epub 2016 Feb 10. J Neurochem. 2016. PMID: 26860955 Review.
-
Molecular mechanisms of α-synuclein and GBA1 in Parkinson's disease.Cell Tissue Res. 2018 Jul;373(1):51-60. doi: 10.1007/s00441-017-2704-y. Epub 2017 Oct 24. Cell Tissue Res. 2018. PMID: 29064079 Free PMC article. Review.
Cited by
-
Lysosomal Storage Disorders Shed Light on Lysosomal Dysfunction in Parkinson's Disease.Int J Mol Sci. 2020 Jul 14;21(14):4966. doi: 10.3390/ijms21144966. Int J Mol Sci. 2020. PMID: 32674335 Free PMC article. Review.
-
Mitochondrial Phenotypes in Parkinson's Diseases-A Focus on Human iPSC-Derived Dopaminergic Neurons.Cells. 2021 Dec 7;10(12):3436. doi: 10.3390/cells10123436. Cells. 2021. PMID: 34943944 Free PMC article. Review.
-
Is Parkinson's disease a lysosomal disorder?Brain. 2018 Aug 1;141(8):2255-2262. doi: 10.1093/brain/awy147. Brain. 2018. PMID: 29860491 Free PMC article.
-
Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson's disease pathogenesis.Acta Neuropathol Commun. 2020 May 6;8(1):63. doi: 10.1186/s40478-020-00935-4. Acta Neuropathol Commun. 2020. PMID: 32375870 Free PMC article. Review.
-
Elevated Dkk1 Mediates Downregulation of the Canonical Wnt Pathway and Lysosomal Loss in an iPSC Model of Neuronopathic Gaucher Disease.Biomolecules. 2020 Dec 3;10(12):1630. doi: 10.3390/biom10121630. Biomolecules. 2020. PMID: 33287247 Free PMC article.
References
-
- Aflaki E, Stubblefield BK, Maniwang E, Lopez G, Moaven N, Goldin E, Marugan J, Patnaik S, Dutra A, Southall N, Zheng W, Tayebi N, Sidransky E. Macrophage models of Gaucher disease for evaluating disease pathogenesis and candidate drugs. Sci Transl Med. 2014;6:240ra73. doi: 10.1126/scitranslmed.3008659. - DOI - PMC - PubMed
-
- Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, Mazzoni P, Pauciulo MW, Nichols WC, Gan-Or Z, Rouleau GA, Chung WK, Wolf P, Oliva P, Keutzer J, Marder K, Zhang X. Glucocerebrosidase activity in Parkinson's disease with and without GBA mutations. Brain. 2015;138:2648–2658. doi: 10.1093/brain/awv179. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous