

Ethanol-Induced Motor Impairment Mediated by Inhibition of α7 Nicotinic Receptors
- PMID: 27445152
- PMCID: PMC4951579
- DOI: 10.1523/JNEUROSCI.0154-16.2016
Ethanol-Induced Motor Impairment Mediated by Inhibition of α7 Nicotinic Receptors
Abstract
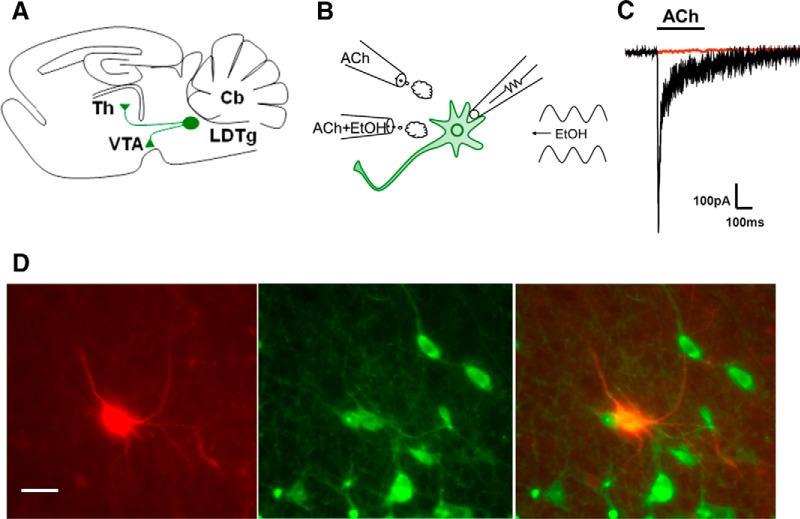
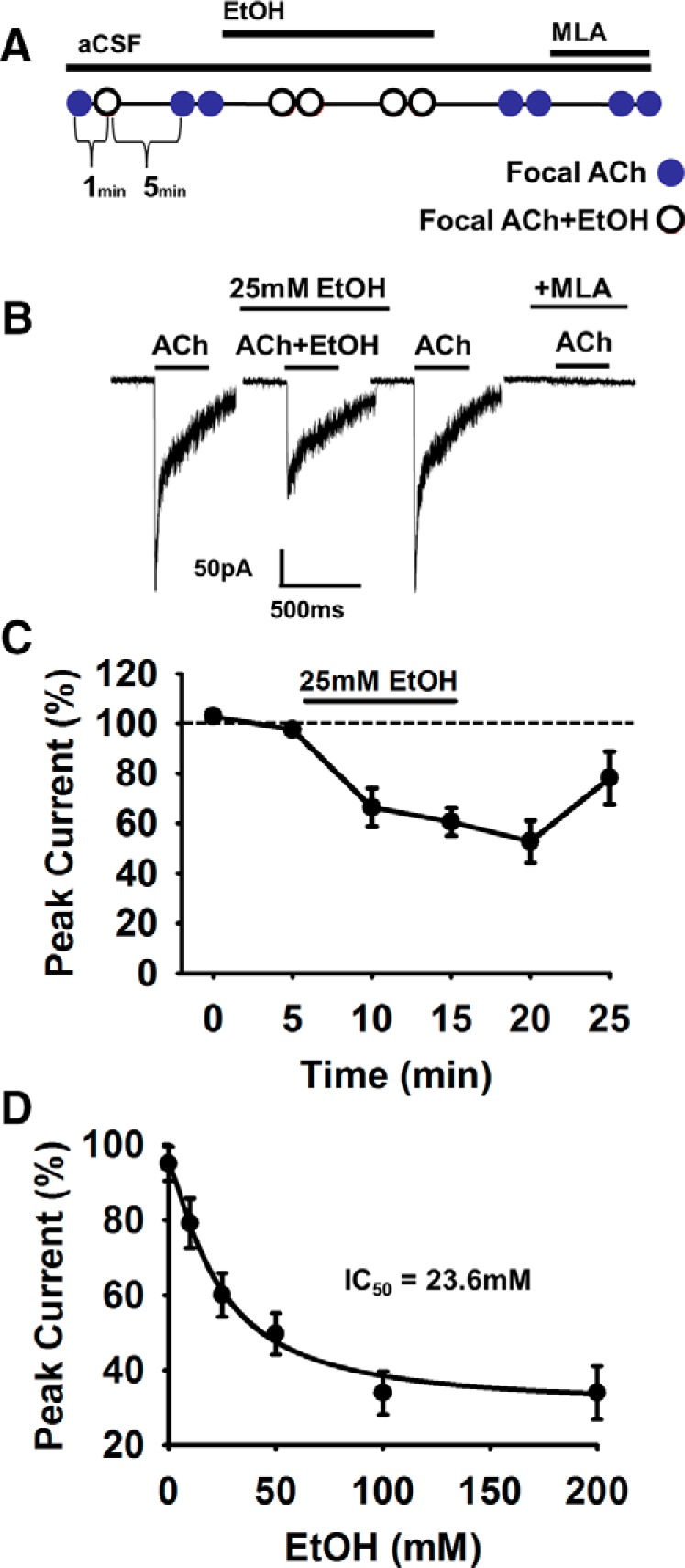
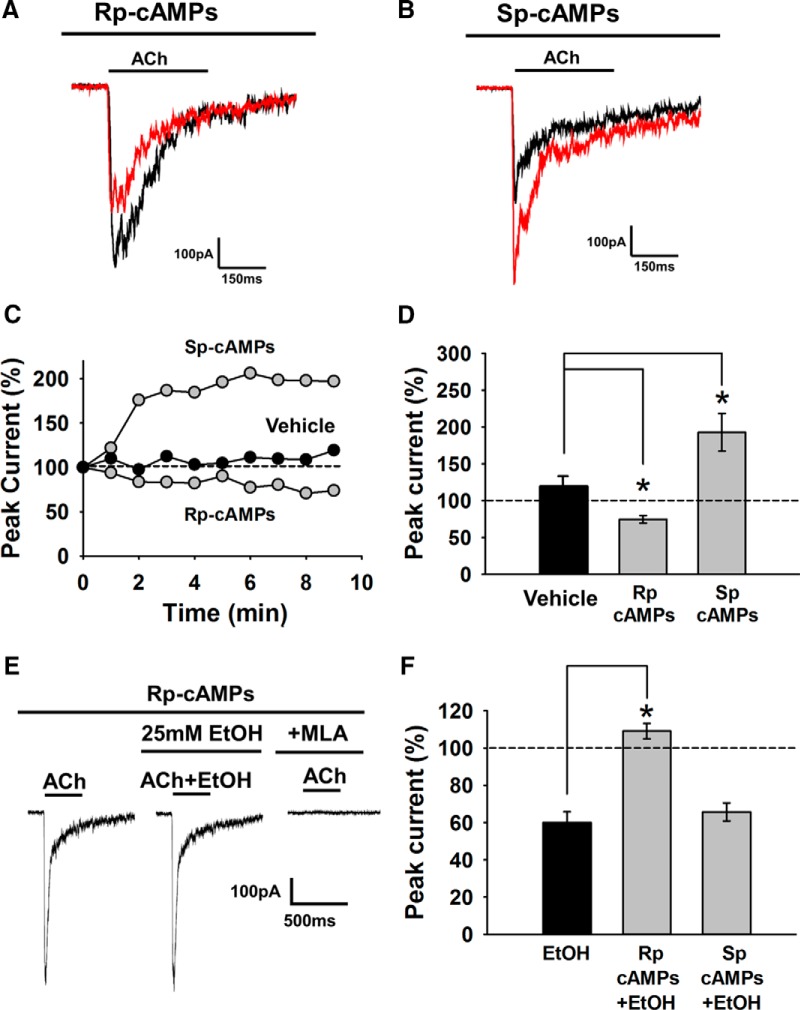
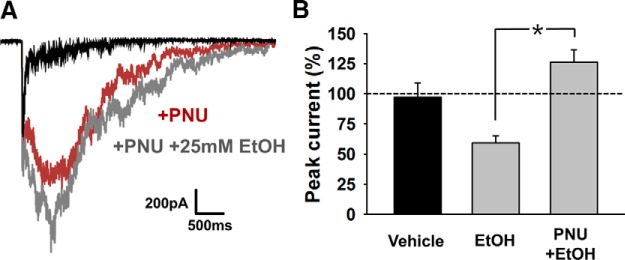
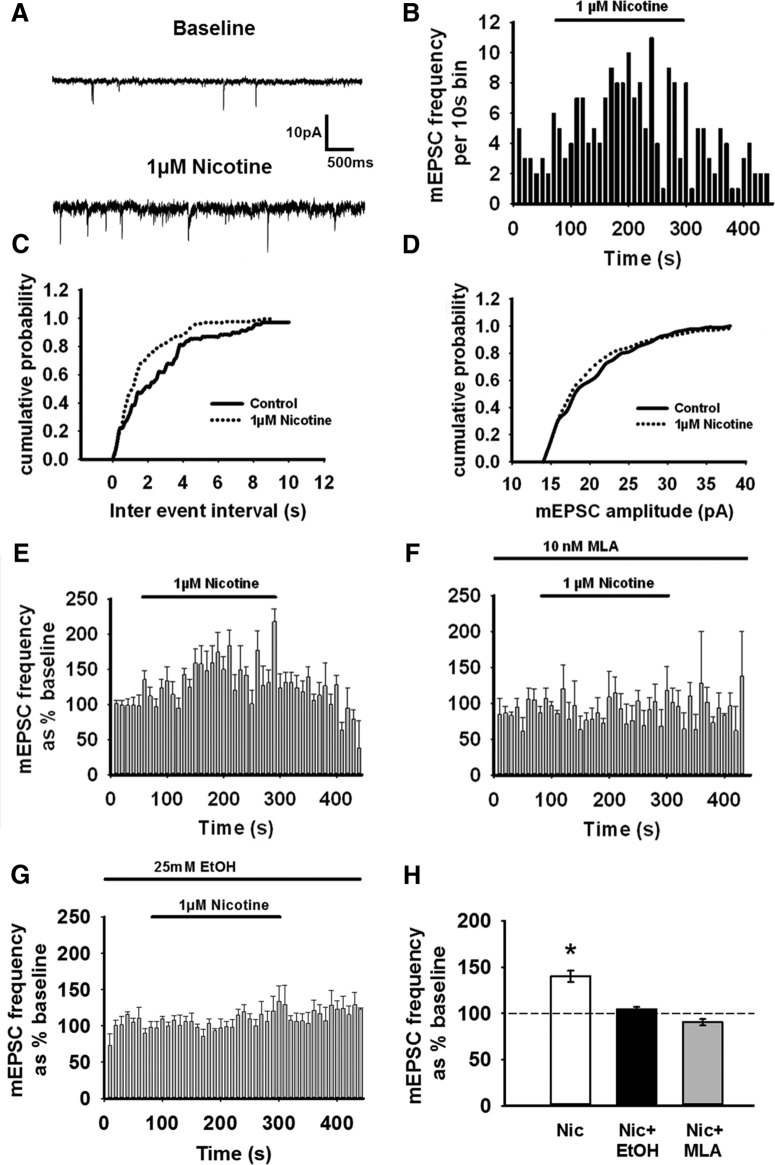
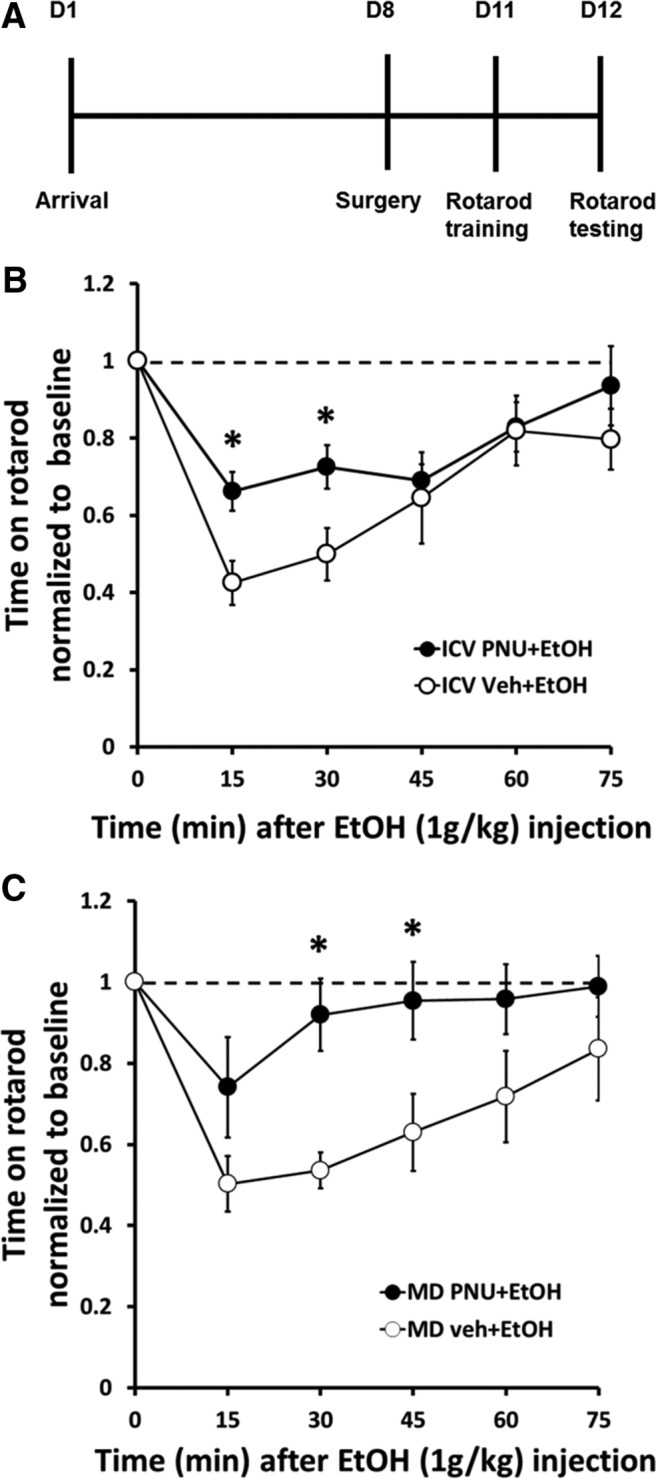
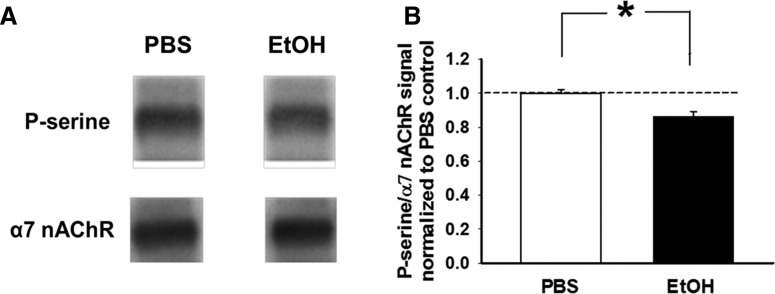
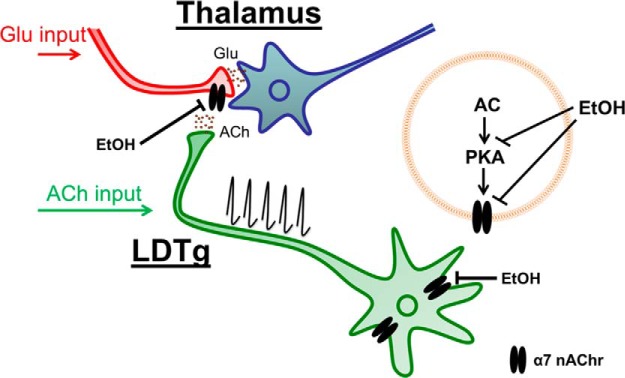
Nicotine and ethanol (EtOH) are among the most widely co-abused substances, and nicotinic acetylcholine receptors (nAChRs) contribute to the behavioral effects of both drugs. Along with their role in addiction, nAChRs also contribute to motor control circuitry. The α7 nAChR subtype is highly expressed in the laterodorsal tegmental nucleus (LDTg), a brainstem cholinergic center that contributes to motor performance through its projections to thalamic motor relay centers, including the mediodorsal thalamus. We demonstrate that EtOH concentrations just above the legal limits for intoxication in humans can inhibit α7 nAChRs in LDTg neurons from rats. This EtOH-induced inhibition is mediated by a decrease in cAMP/PKA signaling. The α7 nAChR-positive allosteric modulator PNU120596 [N-(5-chloro-2,4-dimethoxyphenyl)-N'-(5-methyl-3-isoxazolyl)-urea], which interferes with receptor desensitization, completely eliminated EtOH modulation of these receptors. These data suggest that EtOH inhibits α7 responses through a PKA-dependent enhancement of receptor desensitization. EtOH also inhibited the effects of nicotine at presynaptic α7 nAChRs on glutamate terminals in the mediodorsal thalamus. In vivo administration of PNU120596 either into the cerebral ventricles or directly into the mediodorsal thalamus attenuated EtOH-induced motor impairment. Thus, α7 nAChRs are likely important mediators of the motor impairing effects of moderate EtOH consumption.
Significance statement: The motor-impairing effects of ethanol contribute to intoxication-related injury and death. Here we explore the cellular and neural circuit mechanisms underlying ethanol-induced motor impairment. Physiologically relevant concentrations of ethanol inhibit activity of a nicotinic receptor subtype that is expressed in brain areas associated with motor control. That receptor inhibition is mediated by decreased receptor phosphorylation, suggesting an indirect modulation of cell signaling pathways to achieve the physiological effects.
Keywords: aversion; ethanol; motor impairment; nicotine; protein kinase A; rotarod.
Copyright © 2016 the authors 0270-6474/16/367768-11$15.00/0.
Figures
References
-
- Aistrup GL, Marszalec W, Narahashi T. Ethanol modulation of nicotinic acetylcholine receptor currents in cultured cortical neurons. Mol Pharmacol. 1999;55:39–49. - PubMed
-
- Azam L, Winzer-Serhan U, Leslie FM. Co-expression of alpha7 and beta2 nicotinic acetylcholine receptor subunit mRNAs within rat brain cholinergic neurons. Neuroscience. 2003;119:965–977. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources