

Protection of Primary Dopaminergic Midbrain Neurons by GPR139 Agonists Supports Different Mechanisms of MPP(+) and Rotenone Toxicity
- PMID: 27445691
- PMCID: PMC4923153
- DOI: 10.3389/fncel.2016.00164
Protection of Primary Dopaminergic Midbrain Neurons by GPR139 Agonists Supports Different Mechanisms of MPP(+) and Rotenone Toxicity
Abstract
The G-protein coupled receptor 139 (GPR139) is expressed specifically in the brain in areas of relevance for motor control. GPR139 function and signal transduction pathways are elusive, and results in the literature are even contradictory. Here, we examined the potential neuroprotective effect of GPR139 agonism in primary culture models of dopaminergic (DA) neuronal degeneration. We find that in vitro GPR139 agonists protected primary mesencephalic DA neurons against 1-methyl-4-phenylpyridinium (MPP(+))-mediated degeneration. Protection was concentration-dependent and could be blocked by a GPR139 antagonist. However, the protection of DA neurons was not found against rotenone or 6-hydroxydopamine (6-OHDA) mediated degeneration. Our results support differential mechanisms of toxicity for those substances commonly used in Parkinson's disease (PD) models and potential for GPR139 agonists in neuroprotection.
Keywords: G protein-coupled receptor; Parkinson’s disease model; apoptosis; cell-based assay; drug screening; neurodegeneration; neuroprotection; neurotoxin.
Figures
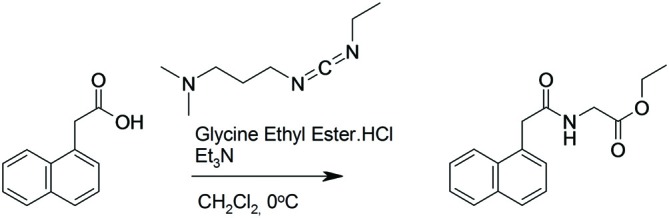

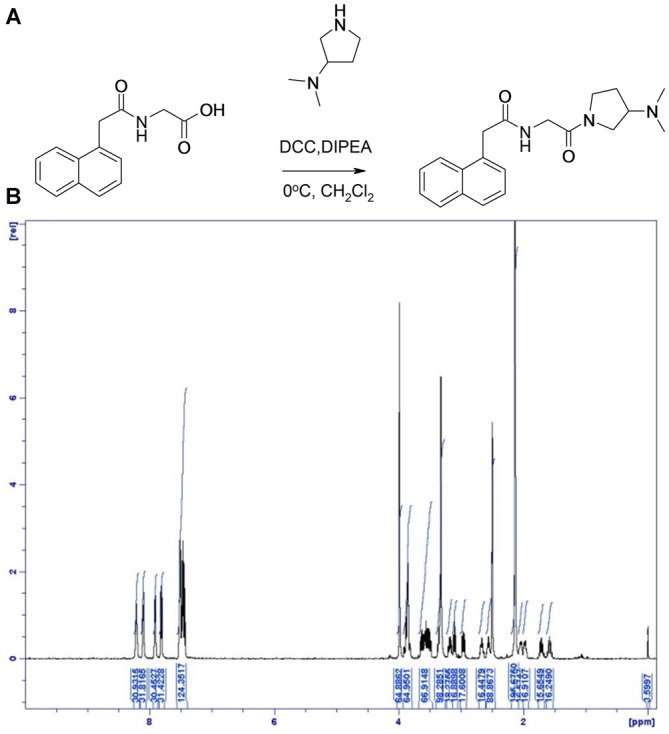
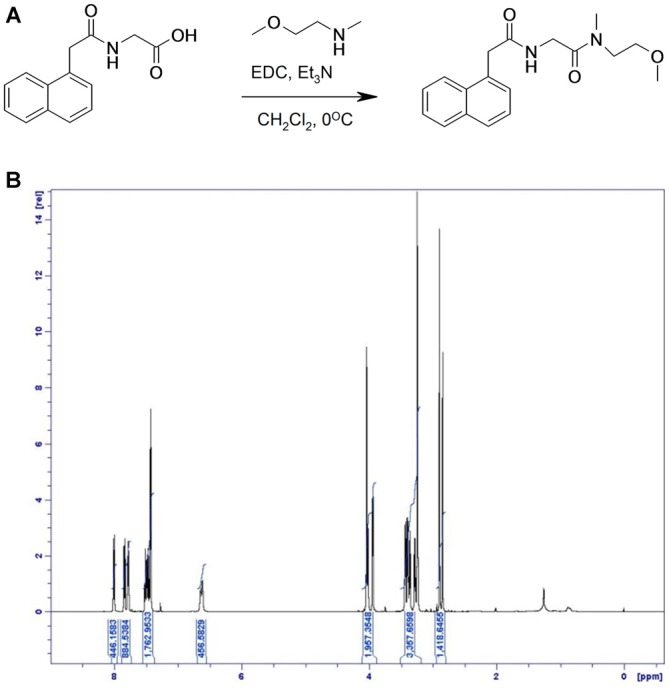

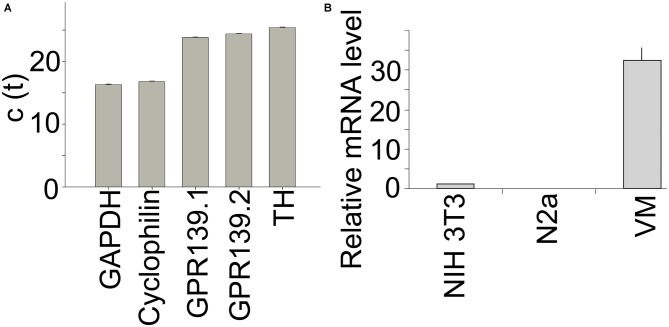
 ), compound 2 (
), compound 2 ( ), or compound 3 (
), or compound 3 ( ), or vehicle (
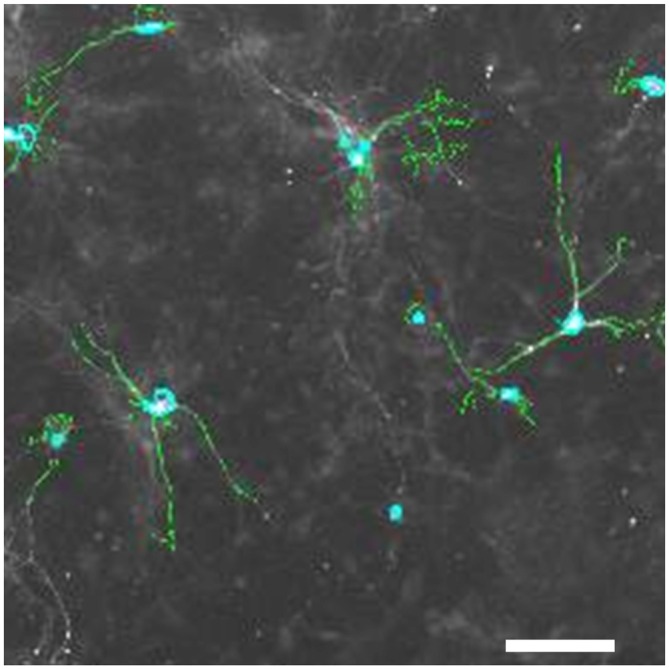
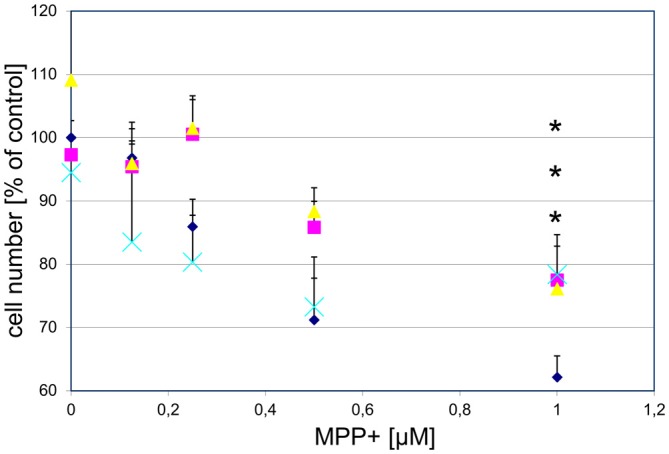
), or vehicle ( ) for 1 h, followed by treatment with the indicated concentrations of MPP+ for 24 h. TH-positive neurons were counted and normalized to numbers under control conditions. At 1 μM MPP+, protection by all three different agonists was significant (*p ≤ 0.05). Each data point is calculated from 12 (0 μM and 1 μM MPP+ concentrations); 4 (0.125 μM); 7 (0.25 μM); or 8 (0.5 μM) independent measurements.
) for 1 h, followed by treatment with the indicated concentrations of MPP+ for 24 h. TH-positive neurons were counted and normalized to numbers under control conditions. At 1 μM MPP+, protection by all three different agonists was significant (*p ≤ 0.05). Each data point is calculated from 12 (0 μM and 1 μM MPP+ concentrations); 4 (0.125 μM); 7 (0.25 μM); or 8 (0.5 μM) independent measurements.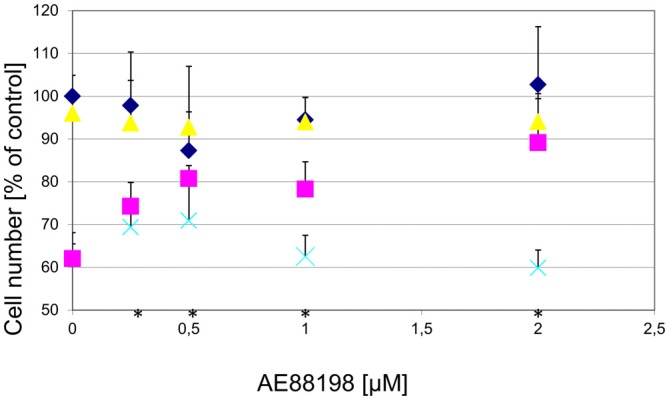 ), or 1 μM MPP+ (); or MPP+ with 10 μM of the antagonist compound 4 (), or with the antagonist compound 4 alone (). The number of TH-positive neurons was determined 24 h later and normalized to control conditions. Statistical significance compared to vehicle treated cells is indicated (*p < 0.05).
), or 1 μM MPP+ (); or MPP+ with 10 μM of the antagonist compound 4 (), or with the antagonist compound 4 alone (). The number of TH-positive neurons was determined 24 h later and normalized to control conditions. Statistical significance compared to vehicle treated cells is indicated (*p < 0.05).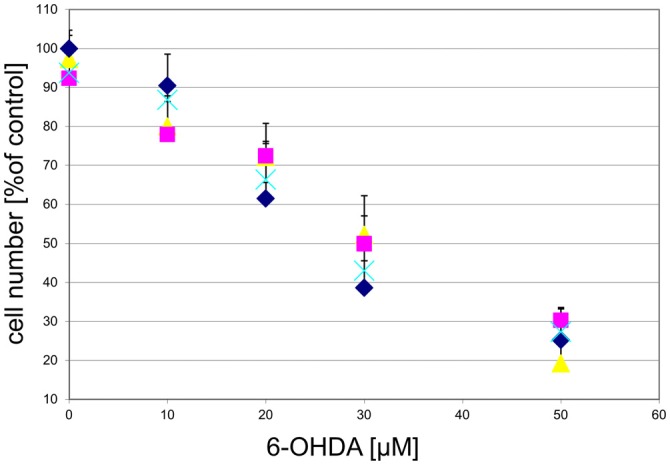 ), compound 2 (), or compound 3 (), or vehicle () for 1 h, followed by treatment with the indicated concentrations of 6-hydroxydopamine (6-OHDA) for 24 h. TH-positive neurons were counted and normalized to control numbers.
), compound 2 (), or compound 3 (), or vehicle () for 1 h, followed by treatment with the indicated concentrations of 6-hydroxydopamine (6-OHDA) for 24 h. TH-positive neurons were counted and normalized to control numbers.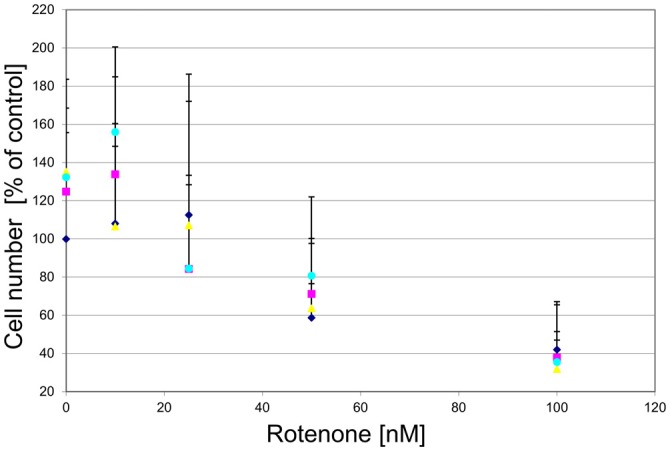 ), compound 2 (), or compound 3 (
), compound 2 (), or compound 3 ( ), or vehicle () for 1 h before exposing them to 0–100 nM of rotenone. Results are average of ± SEM (n = 4 independent experiments). TH-positive neurons were counted and normalized to control numbers (100%). At a given rotenone concentration, none of the values among the treatment groups were significantly different from one another.
), or vehicle () for 1 h before exposing them to 0–100 nM of rotenone. Results are average of ± SEM (n = 4 independent experiments). TH-positive neurons were counted and normalized to control numbers (100%). At a given rotenone concentration, none of the values among the treatment groups were significantly different from one another.References
LinkOut - more resources
Full Text Sources
Other Literature Sources