

Small molecule epigenetic screen identifies novel EZH2 and HDAC inhibitors that target glioblastoma brain tumor-initiating cells
- PMID: 27449082
- PMCID: PMC5312317
- DOI: 10.18632/oncotarget.10661
Small molecule epigenetic screen identifies novel EZH2 and HDAC inhibitors that target glioblastoma brain tumor-initiating cells
Abstract
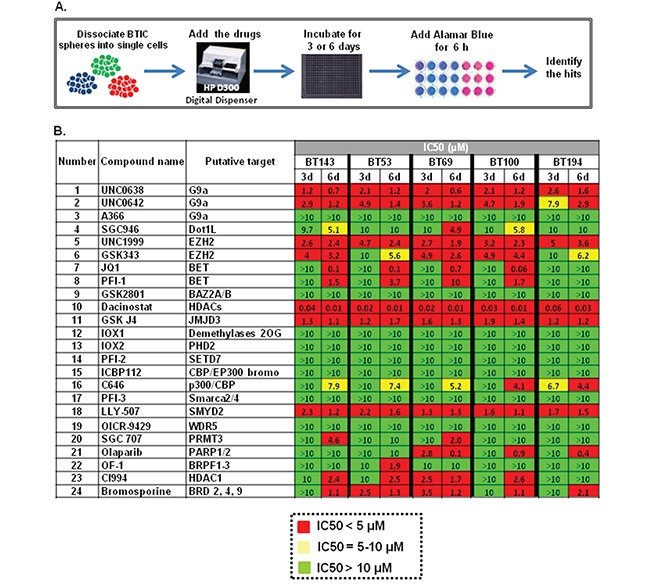
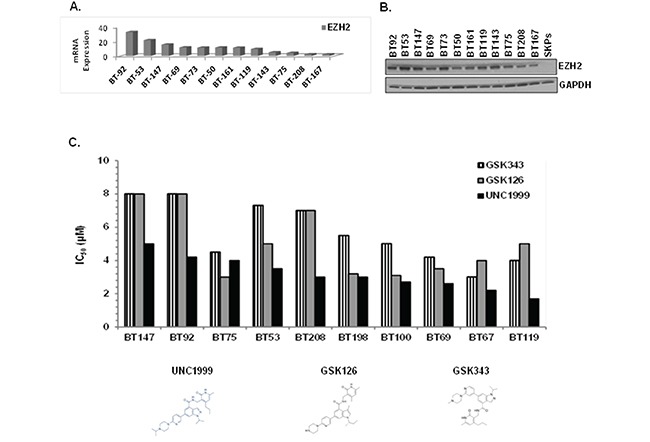
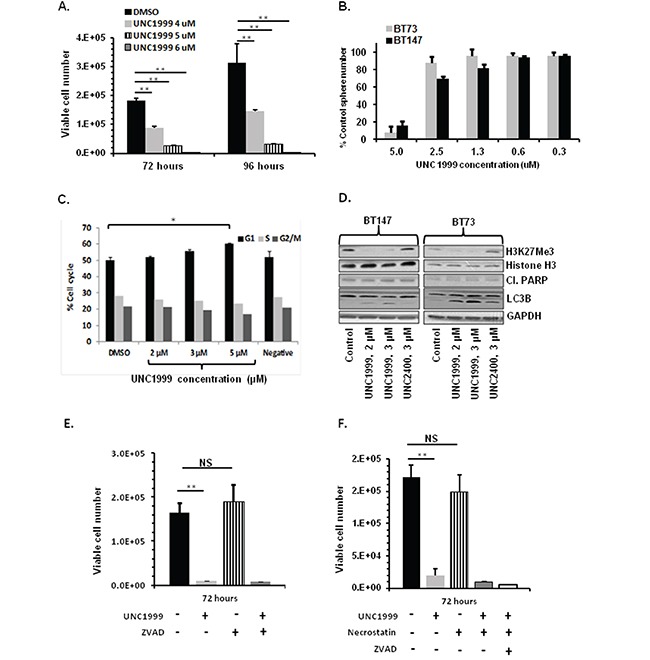
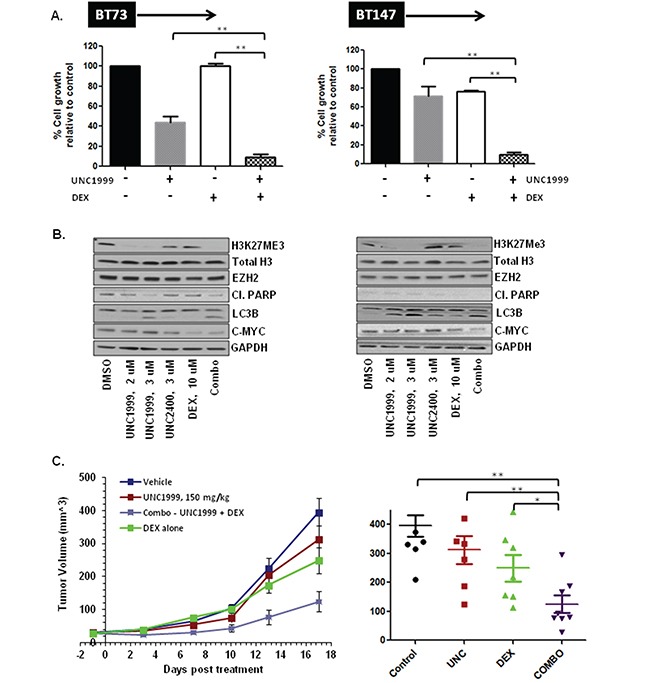
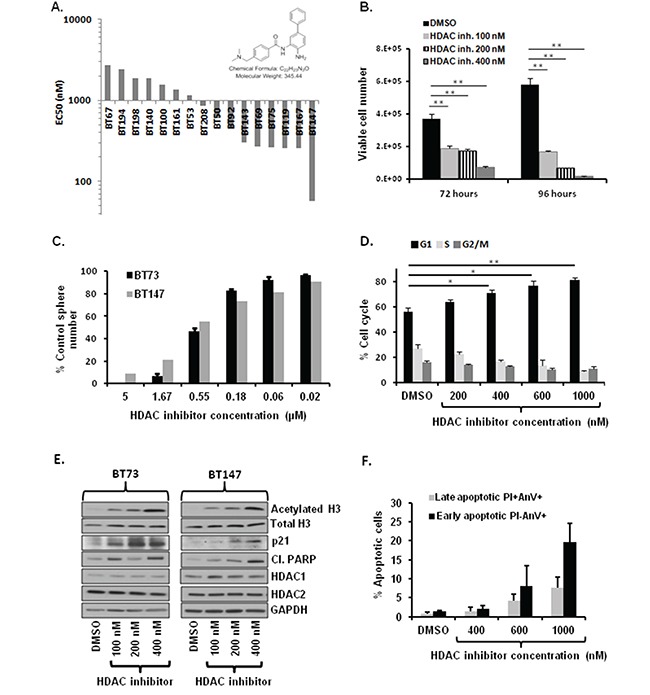
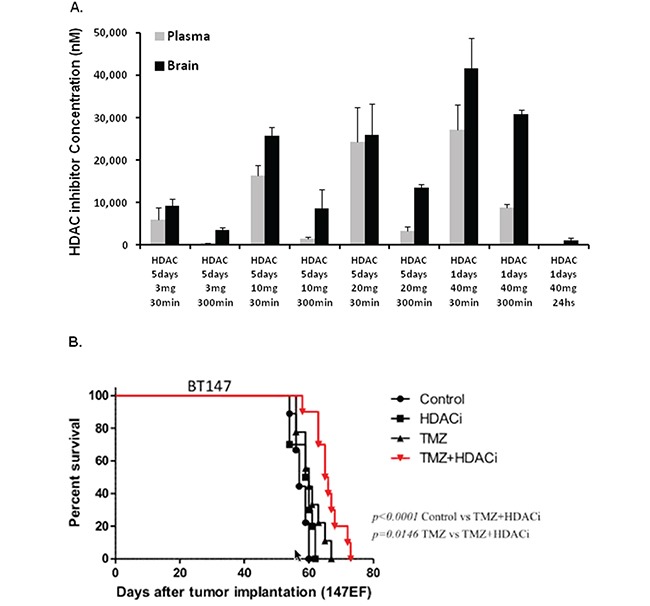
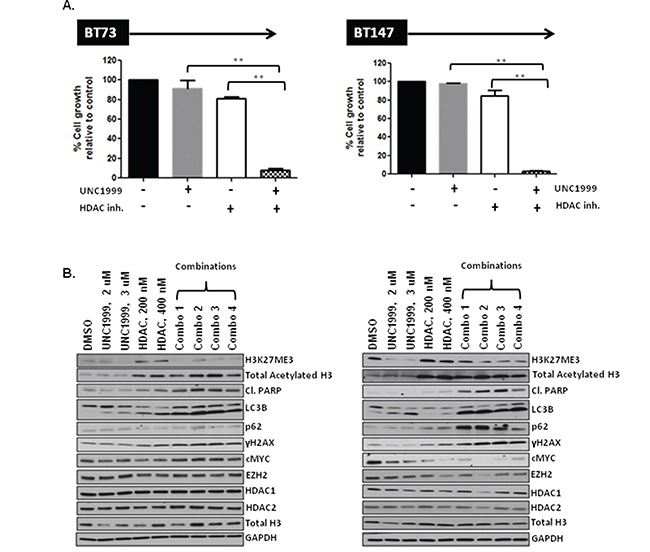
Glioblastoma (GBM) is the most lethal and aggressive adult brain tumor, requiring the development of efficacious therapeutics. Towards this goal, we screened five genetically distinct patient-derived brain-tumor initiating cell lines (BTIC) with a unique collection of small molecule epigenetic modulators from the Structural Genomics Consortium (SGC). We identified multiple hits that inhibited the growth of BTICs in vitro, and further evaluated the therapeutic potential of EZH2 and HDAC inhibitors due to the high relevance of these targets for GBM. We found that the novel SAM-competitive EZH2 inhibitor UNC1999 exhibited low micromolar cytotoxicity in vitro on a diverse collection of BTIC lines, synergized with dexamethasone (DEX) and suppressed tumor growth in vivo in combination with DEX. In addition, a unique brain-penetrant class I HDAC inhibitor exhibited cytotoxicity in vitro on a panel of BTIC lines and extended survival in combination with TMZ in an orthotopic BTIC model in vivo. Finally, a combination of EZH2 and HDAC inhibitors demonstrated synergy in vitro by augmenting apoptosis and increasing DNA damage. Our findings identify key epigenetic modulators in GBM that regulate BTIC growth and survival and highlight promising combination therapies.
Keywords: HDAC inhibitor; UNC1999; drug discovery; epigenetics; glioblastoma.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. - PubMed
-
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
