

Antioxidant Protection of NADPH-Depleted Oligodendrocyte Precursor Cells Is Dependent on Supply of Reduced Glutathione
- PMID: 27449129
- PMCID: PMC4962338
- DOI: 10.1177/1759091416660404
Antioxidant Protection of NADPH-Depleted Oligodendrocyte Precursor Cells Is Dependent on Supply of Reduced Glutathione
Abstract
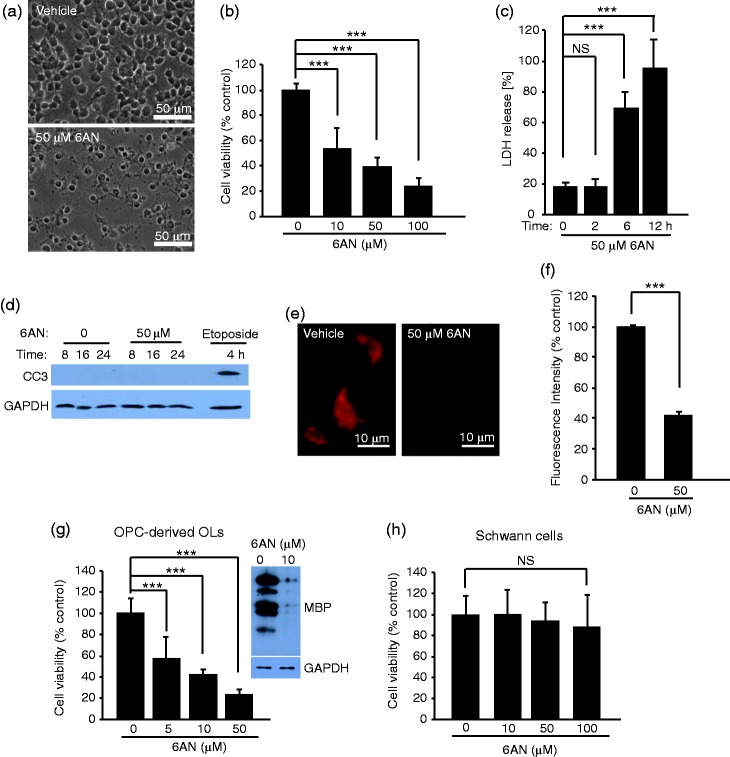
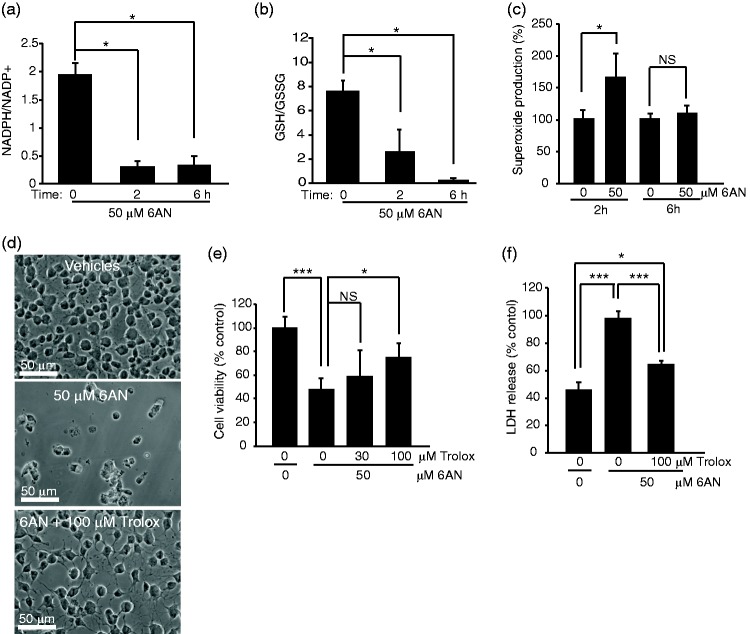
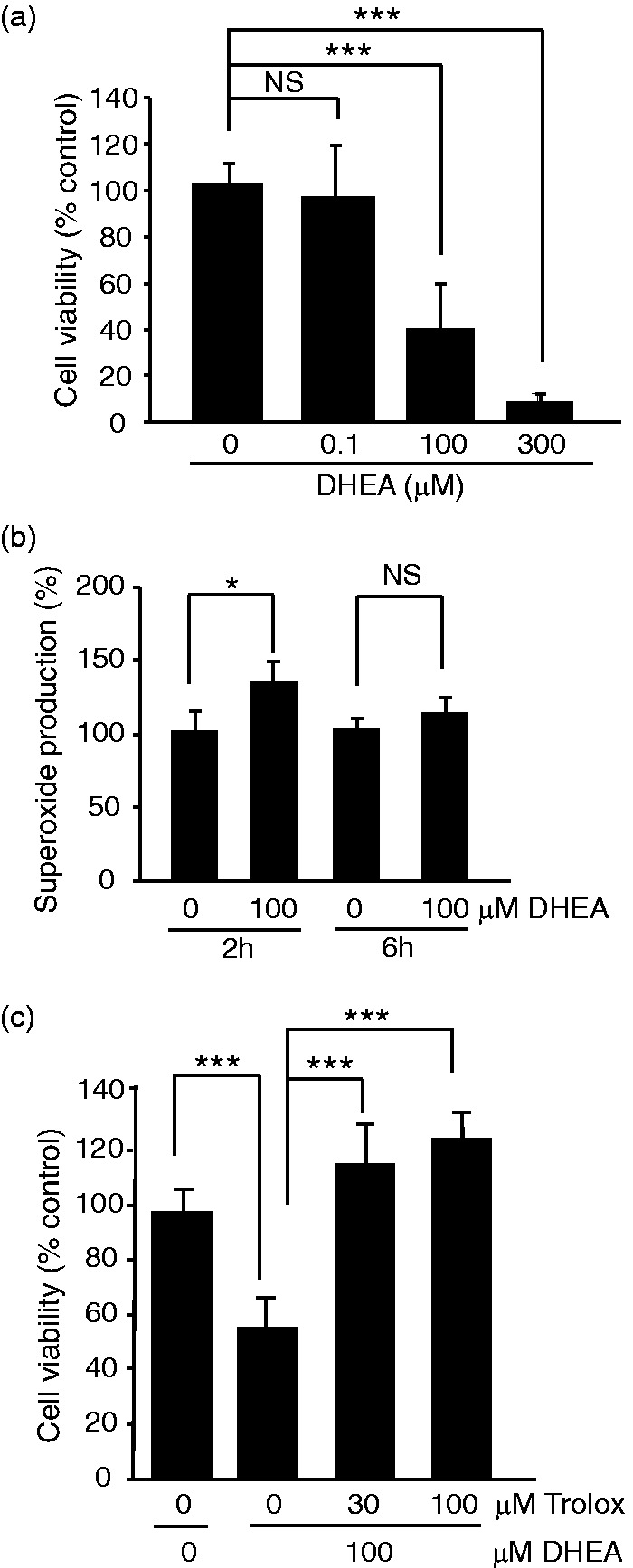
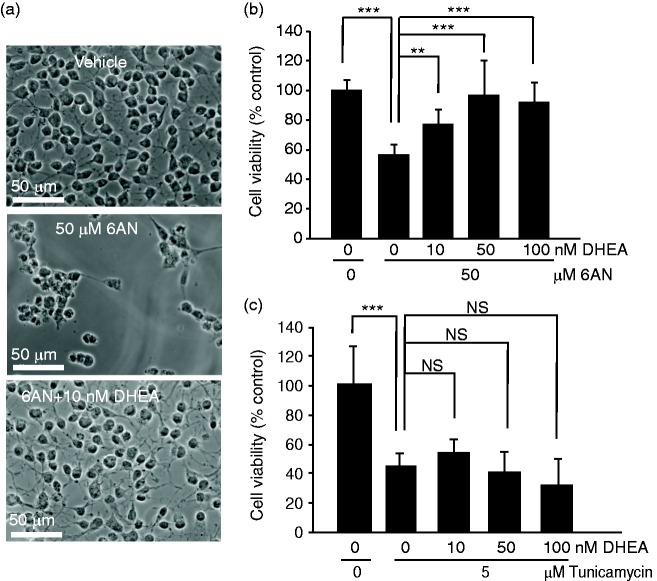
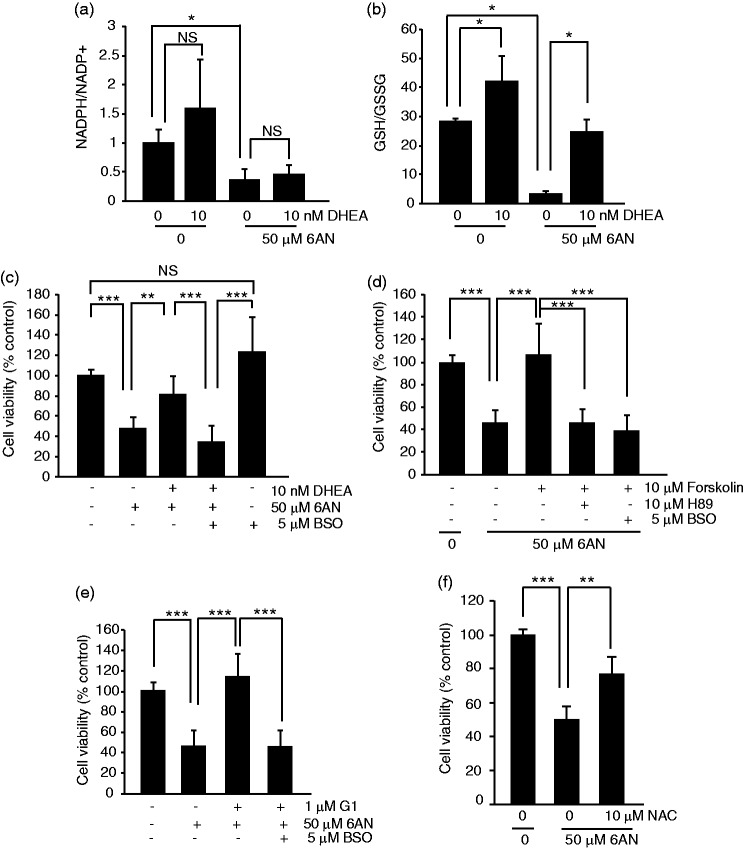
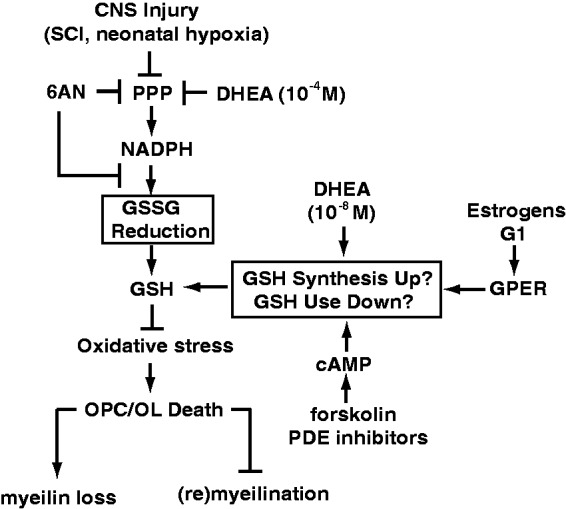
The pentose phosphate pathway is the main source of NADPH, which by reducing oxidized glutathione, contributes to antioxidant defenses. Although oxidative stress plays a major role in white matter injury, significance of NADPH for oligodendrocyte survival has not been yet investigated. It is reported here that the NADPH antimetabolite 6-amino-NADP (6AN) was cytotoxic to cultured adult rat spinal cord oligodendrocyte precursor cells (OPCs) as well as OPC-derived oligodendrocytes. The 6AN-induced necrosis was preceded by increased production of superoxide, NADPH depletion, and lower supply of reduced glutathione. Moreover, survival of NADPH-depleted OPCs was improved by the antioxidant drug trolox. Such cells were also protected by physiological concentrations of the neurosteroid dehydroepiandrosterone (10(-8) M). The protection by dehydroepiandrosterone was associated with restoration of reduced glutathione, but not NADPH, and was sensitive to inhibition of glutathione synthesis. A similar protective mechanism was engaged by the cAMP activator forskolin or the G protein-coupled estrogen receptor (GPER/GPR30) ligand G1. Finally, treatment with the glutathione precursor N-acetyl cysteine reduced cytotoxicity of 6AN. Taken together, NADPH is critical for survival of OPCs by supporting their antioxidant defenses. Consequently, injury-associated inhibition of the pentose phosphate pathway may be detrimental for the myelination or remyelination potential of the white matter. Conversely, steroid hormones and cAMP activators may promote survival of NADPH-deprived OPCs by increasing a NADPH-independent supply of reduced glutathione. Therefore, maintenance of glutathione homeostasis appears as a critical effector mechanism for OPC protection against NADPH depletion and preservation of the regenerative potential of the injured white matter.
Keywords: demyelination; metabolism; oligodendrocytes; oxidative stress; pentose phosphate pathway; white matter.
© The Author(s) 2016.
Figures
Similar articles
-
Developmental differences in HO-induced oligodendrocyte cell death: role of glutathione, mitogen-activated protein kinases and caspase 3.J Neurochem. 2004 Jul;90(2):392-404. doi: 10.1111/j.1471-4159.2004.02488.x. J Neurochem. 2004. PMID: 15228596
-
A functional role of NMDA receptor in regulating the differentiation of oligodendrocyte precursor cells and remyelination.Glia. 2013 May;61(5):732-49. doi: 10.1002/glia.22469. Epub 2013 Feb 26. Glia. 2013. PMID: 23440860
-
CNTF promotes the survival and differentiation of adult spinal cord-derived oligodendrocyte precursor cells in vitro but fails to promote remyelination in vivo.Exp Neurol. 2007 Mar;204(1):485-9. doi: 10.1016/j.expneurol.2006.12.013. Epub 2006 Dec 20. Exp Neurol. 2007. PMID: 17274982 Free PMC article.
-
Oligodendrocyte-protection and remyelination post-spinal cord injuries: a review.Prog Neurobiol. 2012 Mar;96(3):322-39. doi: 10.1016/j.pneurobio.2012.01.008. Epub 2012 Jan 28. Prog Neurobiol. 2012. PMID: 22307058 Review.
-
Cellular reducing equivalents and oxidative stress.Free Radic Biol Med. 1994 Jul;17(1):65-75. doi: 10.1016/0891-5849(94)90008-6. Free Radic Biol Med. 1994. PMID: 7959167 Review.
Cited by
-
From BBB to PPP: Bioenergetic requirements and challenges for oligodendrocytes in health and disease.J Neurochem. 2025 Jan;169(1):e16219. doi: 10.1111/jnc.16219. Epub 2024 Sep 10. J Neurochem. 2025. PMID: 39253904 Free PMC article. Review.
-
Investigating the antioxidant potential and mechanism of a hydrazide bioactive component of garlic: insights from density functional theory calculations, drug-likeness and molecular docking studies.Appl Biochem Biotechnol. 2025 Feb;197(2):847-872. doi: 10.1007/s12010-024-05051-w. Epub 2024 Sep 18. Appl Biochem Biotechnol. 2025. PMID: 39292337
-
Cell of all trades: oligodendrocyte precursor cells in synaptic, vascular, and immune function.Genes Dev. 2021 Feb 1;35(3-4):180-198. doi: 10.1101/gad.344218.120. Genes Dev. 2021. PMID: 33526585 Free PMC article. Review.
-
The AMPK activator metformin improves recovery from demyelination by shifting oligodendrocyte bioenergetics and accelerating OPC differentiation.Front Cell Neurosci. 2023 Oct 12;17:1254303. doi: 10.3389/fncel.2023.1254303. eCollection 2023. Front Cell Neurosci. 2023. PMID: 37904733 Free PMC article.
-
The role of oligodendrocytes and their progenitors on neural interface technology: A novel perspective on tissue regeneration and repair.Biomaterials. 2018 Nov;183:200-217. doi: 10.1016/j.biomaterials.2018.08.046. Epub 2018 Aug 22. Biomaterials. 2018. PMID: 30172245 Free PMC article. Review.
References
-
- Acs P., Kipp M., Norkute A., Johann S., Clarner T., Braun A., Beyer C. (2009) 17beta-estradiol and progesterone prevent cuprizone provoked demyelination of corpus callosum in male mice. Glia 57: 807–814. - PubMed
-
- Aragno M., Parola S., Brignardello E., Mauro A., Tamagno E., Manti R., Boccuzzi G. (2000) Dehydroepiandrosterone prevents oxidative injury induced by transient ischemia/reperfusion in the brain of diabetic rats. Diabetes 49: 1924–1931. - PubMed
-
- Bartnik B. L., Sutton R. L., Fukushima M., Harris N. G., Hovda D. A., Lee S. M. (2005) Upregulation of pentose phosphate pathway and preservation of tricarboxylic acid cycle flux after experimental brain injury. Journal of Neurotrauma 22: 1052–1065. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical