

Identification of a Large DNAJB2 Deletion in a Family with Spinal Muscular Atrophy and Parkinsonism
- PMID: 27449489
- PMCID: PMC5375037
- DOI: 10.1002/humu.23055
Identification of a Large DNAJB2 Deletion in a Family with Spinal Muscular Atrophy and Parkinsonism
Abstract
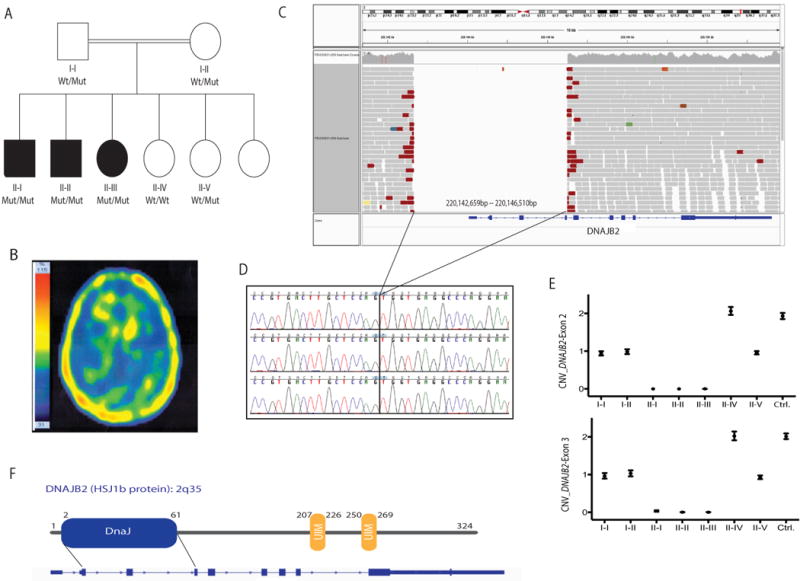
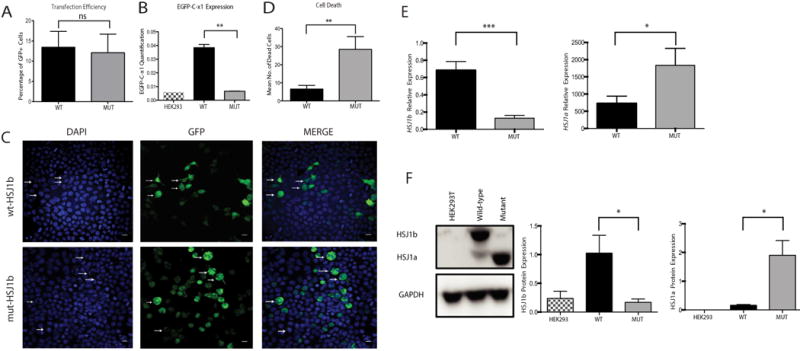
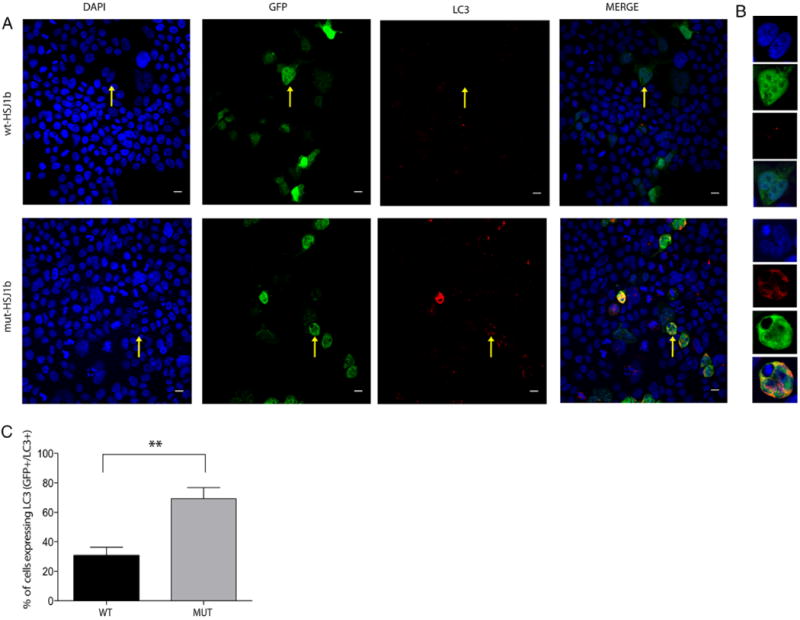
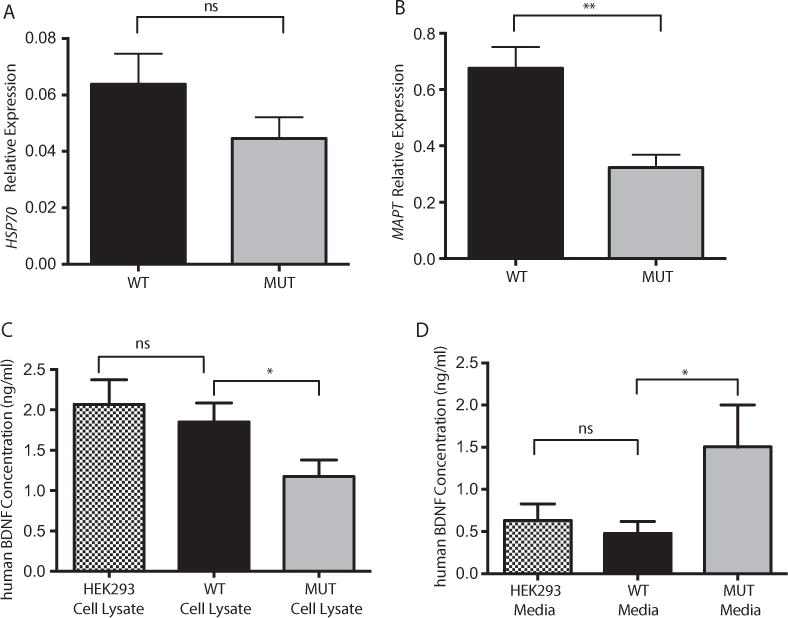
In this study, we described the identification of a large DNAJB2 (HSJ1) deletion in a family with recessive spinal muscular atrophy and Parkinsonism. After performing homozygosity mapping and whole genome sequencing, we identified a 3.8 kb deletion, spanning the entire DnaJ domain of the HSJ1 protein, as the disease-segregating mutation. By performing functional assays, we showed that HSJ1b-related DnaJ domain deletion leads to loss of HSJ1b mRNA and protein levels, increased HSJ1a mRNA and protein expressions, increased cell death, protein aggregation, and enhanced autophagy. Given the role of HSJ1 proteins in the degradation of misfolded proteins, we speculated that enhanced autophagy might be promoted by the elevated HSJ1a expression seen in HSJ1b-deficient cells. We also observed a significant reduction in both tau and brain-derived neurotrophic factor levels, which may explain the dopaminergic deficits seen in one of the affected siblings. We concluded that HSJ1b deficiency leads to a complex neurological phenotype, possibly due to the accumulation of misfolded proteins, caused by the lack of the DnaJ domain activity. We thus expand the phenotypic and genotypic spectrums associated with DNAJB2 disease and suggest relevant disease-associated mechanisms.
Keywords: DNAJB2; DnaJ domain deletion; Parkinsonism; WGS; spinal muscular atrophy.
© 2016 WILEY PERIODICALS, INC.
Conflict of interest statement
Figures
References
-
- Arancio O, Chao MV. Neurotrophins, synaptic plasticity and dementia. Curr Opin Neurobiol. 2007;17:325–330. - PubMed
-
- Blumen SC, Astord S, Robin V, Vignaud L, Toumi N, Cieslik A, Achiron A, Carasso RL, Gurevich M, Braverman I, et al. A rare recessive distal hereditary motor neuropathy with HSJ1 chaperone mutation. Ann Neurol. 2012;71:509–519. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
