

Mitochondrial lipids in neurodegeneration
- PMID: 27449929
- PMCID: PMC5203858
- DOI: 10.1007/s00441-016-2463-1
Mitochondrial lipids in neurodegeneration
Abstract
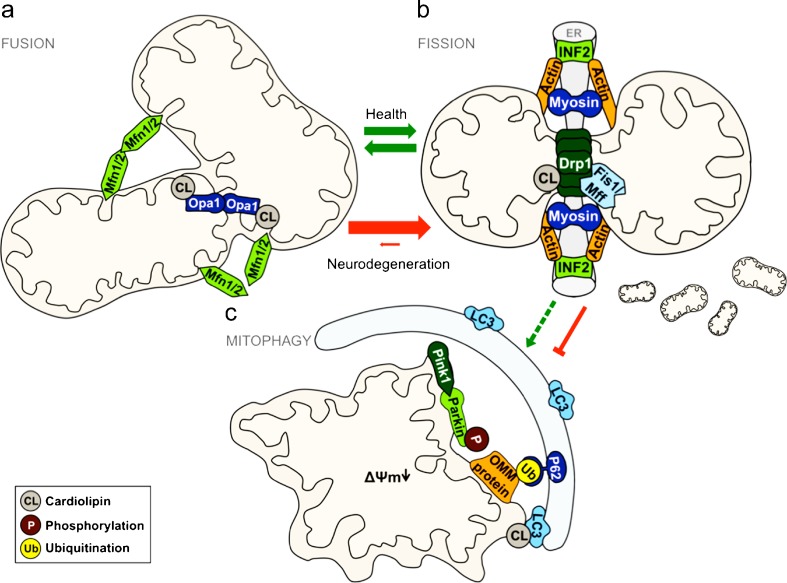
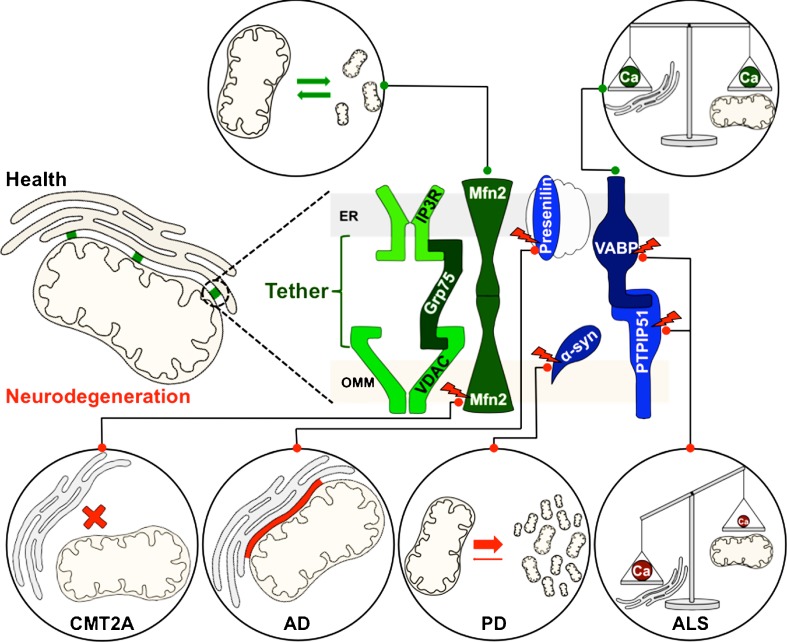
Mitochondrial dysfunction is a common feature of many neurodegenerative diseases, including proteinopathies such as Alzheimer's or Parkinson's disease, which are characterized by the deposition of aggregated proteins in the form of insoluble fibrils or plaques. The distinct molecular processes that eventually result in mitochondrial dysfunction during neurodegeneration are well studied but still not fully understood. However, defects in mitochondrial fission and fusion, mitophagy, oxidative phosphorylation and mitochondrial bioenergetics have been linked to cellular demise. These processes are influenced by the lipid environment within mitochondrial membranes as, besides membrane structure and curvature, recruitment and activity of different proteins also largely depend on the respective lipid composition. Hence, the interaction of neurotoxic proteins with certain lipids and the modification of lipid composition in different cell compartments, in particular mitochondria, decisively impact cell death associated with neurodegeneration. Here, we discuss the relevance of mitochondrial lipids in the pathological alterations that result in neuronal demise, focussing on proteinopathies.
Keywords: Lipids; Mitochondria; Mitochondria-associated membranes; Mitochondrial dynamics; Neurodegeneration.
Figures
References
-
- Alavi MV, Bette S, Schimpf S, Schuettauf F, Schraermeyer U, Wehrl HF, Ruttiger L, Beck SC, Tonagel F, Pichler BJ, Knipper M, Peters T, Laufs J, Wissinger B. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain. 2007;130:1029–1042. doi: 10.1093/brain/awm005. - DOI - PubMed
-
- Ardail D, Privat JP, Egret-Charlier M, Levrat C, Lerme F, Louisot P. Mitochondrial contact sites. Lipid composition and dynamics. J Biol Chem. 1990;265:18797–18802. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
