

CRISPR/Cas9 Screens Reveal Requirements for Host Cell Sulfation and Fucosylation in Bacterial Type III Secretion System-Mediated Cytotoxicity
- PMID: 27453484
- PMCID: PMC4982808
- DOI: 10.1016/j.chom.2016.06.010
CRISPR/Cas9 Screens Reveal Requirements for Host Cell Sulfation and Fucosylation in Bacterial Type III Secretion System-Mediated Cytotoxicity
Abstract
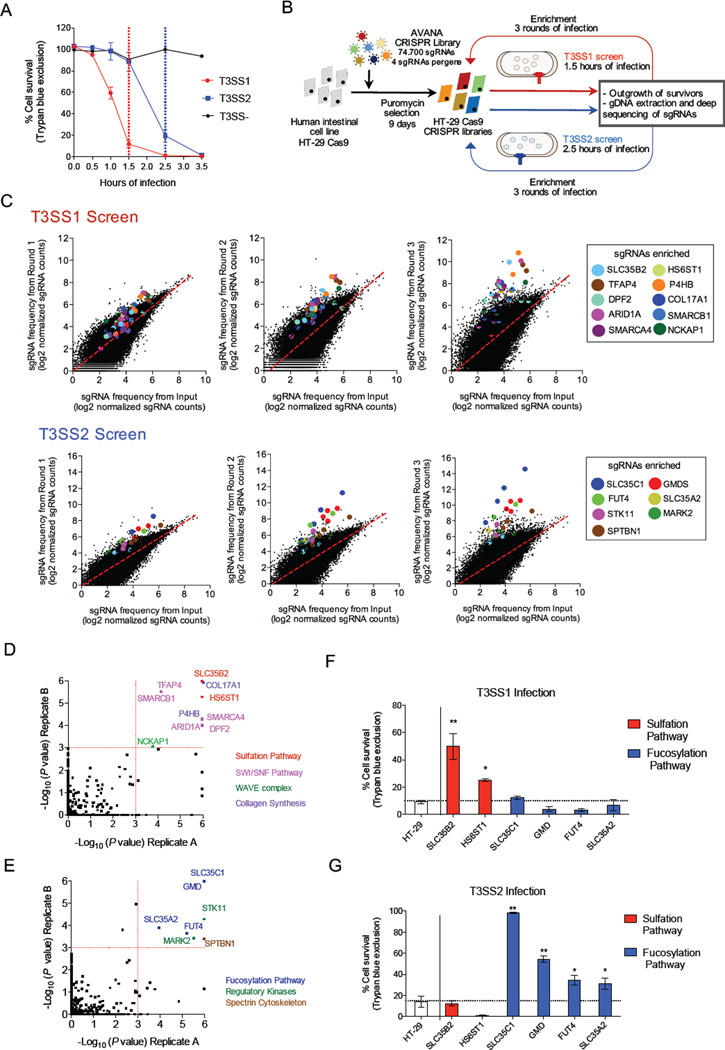
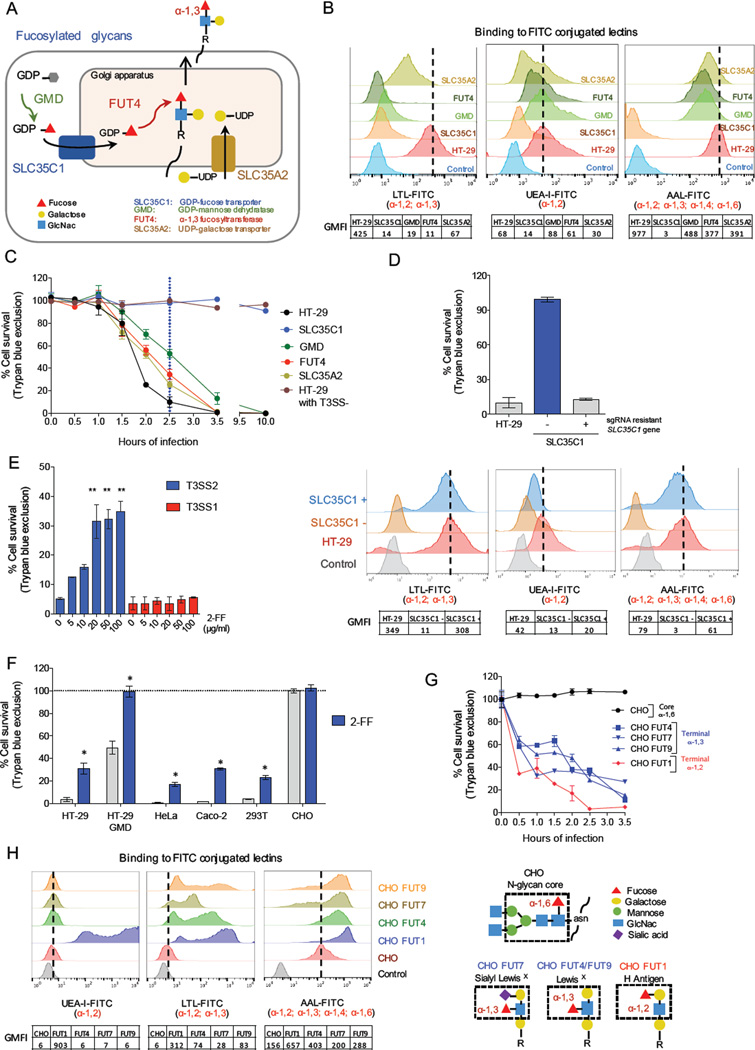
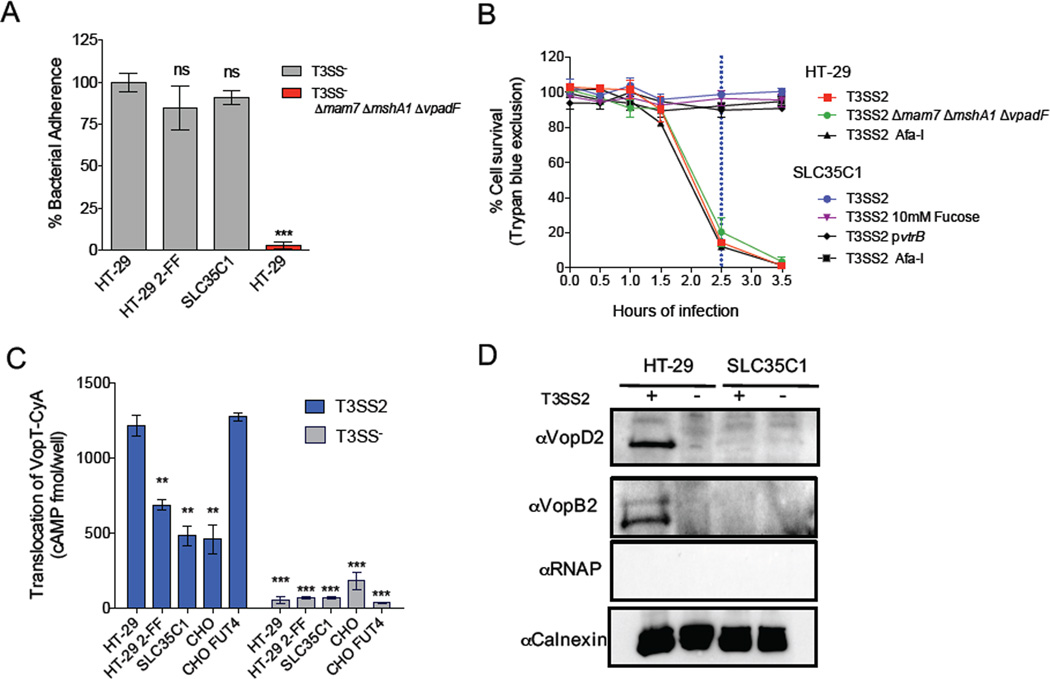
Type III secretion systems (T3SSs) inject bacterial effector proteins into host cells and underlie the virulence of many gram-negative pathogens. Studies have illuminated bacterial factors required for T3SS function, but the required host processes remain largely undefined. We coupled CRISPR/Cas9 genome editing technology with the cytotoxicity of two Vibrio parahaemolyticus T3SSs (T3SS1 and T3SS2) to identify human genome disruptions conferring resistance to T3SS-dependent cytotoxicity. We identity non-overlapping genes required for T3SS1- and T3SS2-mediated cytotoxicity. Genetic ablation of cell surface sulfation reduces bacterial adhesion and thereby alters the kinetics of T3SS1-mediated cytotoxicity. Cell surface fucosylation is required for T3SS2-dependent killing, and genetic inhibition of fucosylation prevents membrane insertion of the T3SS2 translocon complex. These findings reveal the importance of ubiquitous surface modifications for T3SS function, potentially explaining the broad tropism of V. parahaemolyticus, and highlight the utility of genome-wide CRISPR/Cas9 screens to discover processes underlying host-pathogen interactions.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures



Comment in
-
Unhealthy Sugars in Bacterial Cytotoxicity.Cell Host Microbe. 2016 Aug 10;20(2):126-8. doi: 10.1016/j.chom.2016.07.014. Cell Host Microbe. 2016. PMID: 27512898
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
