

Computational investigation of cold denaturation in the Trp-cage miniprotein
- PMID: 27457961
- PMCID: PMC4987839
- DOI: 10.1073/pnas.1607500113
Computational investigation of cold denaturation in the Trp-cage miniprotein
Abstract
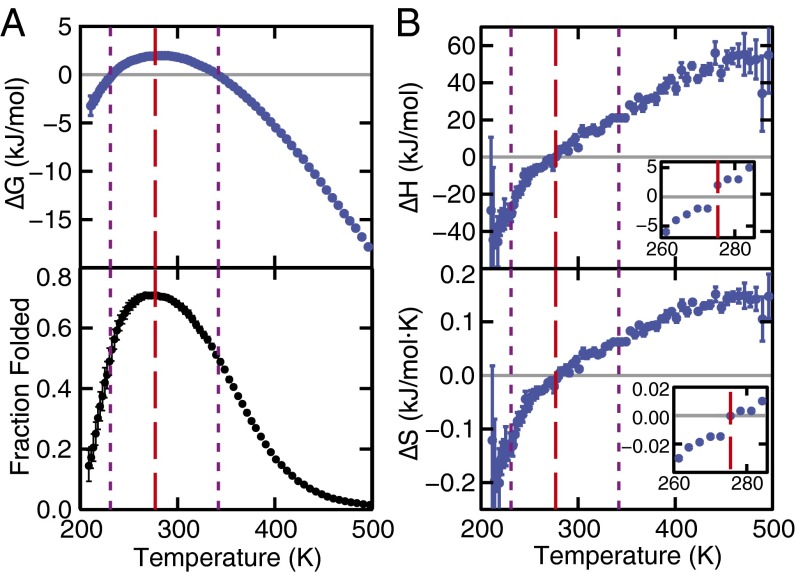
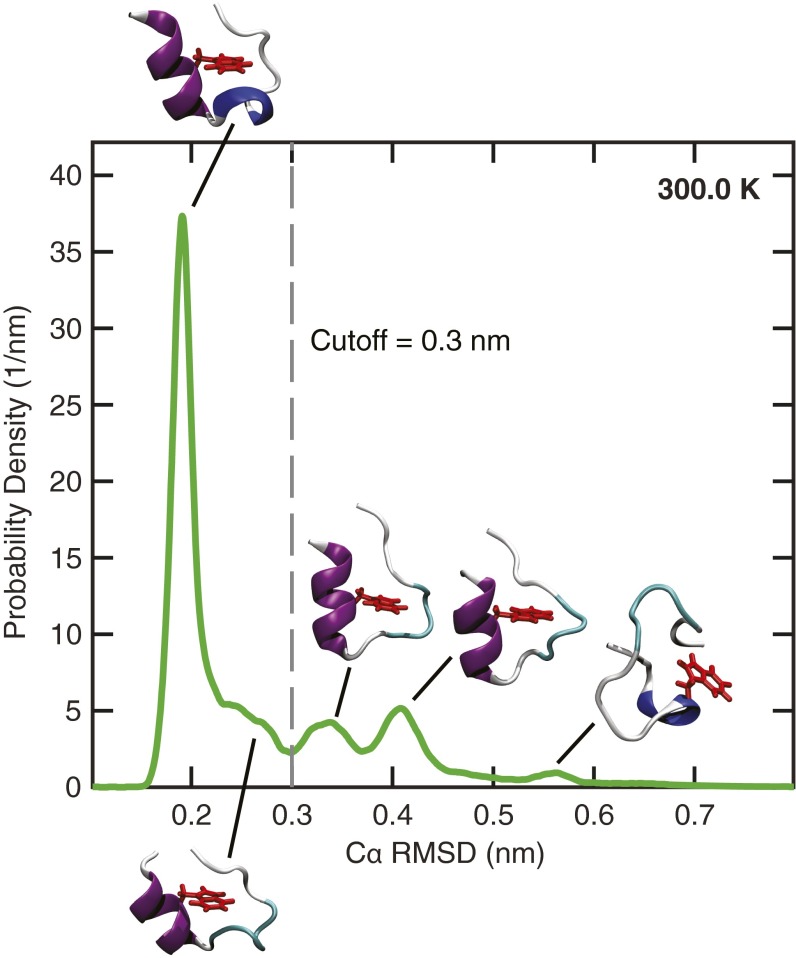
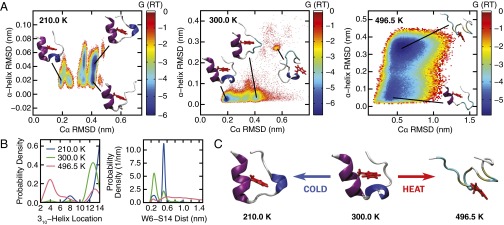
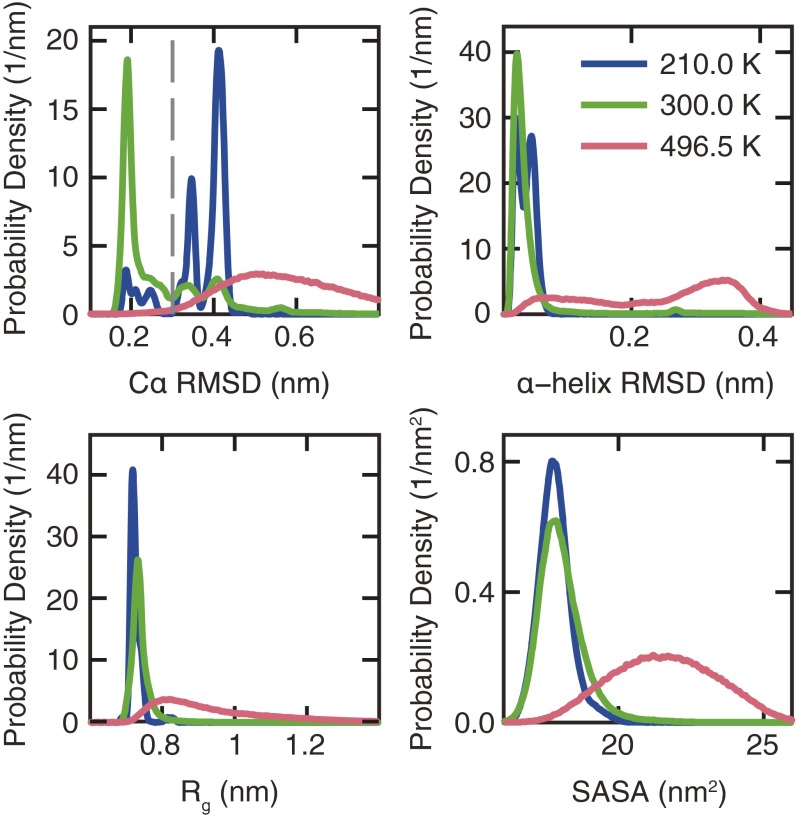
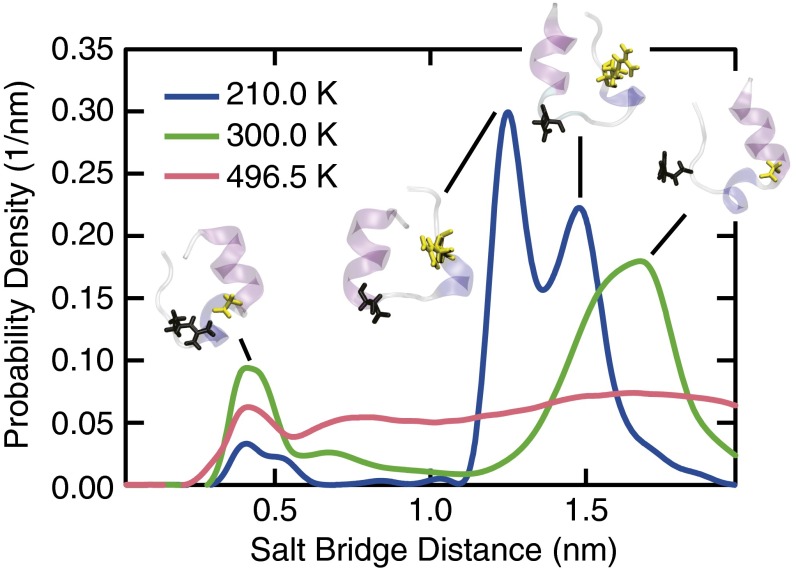
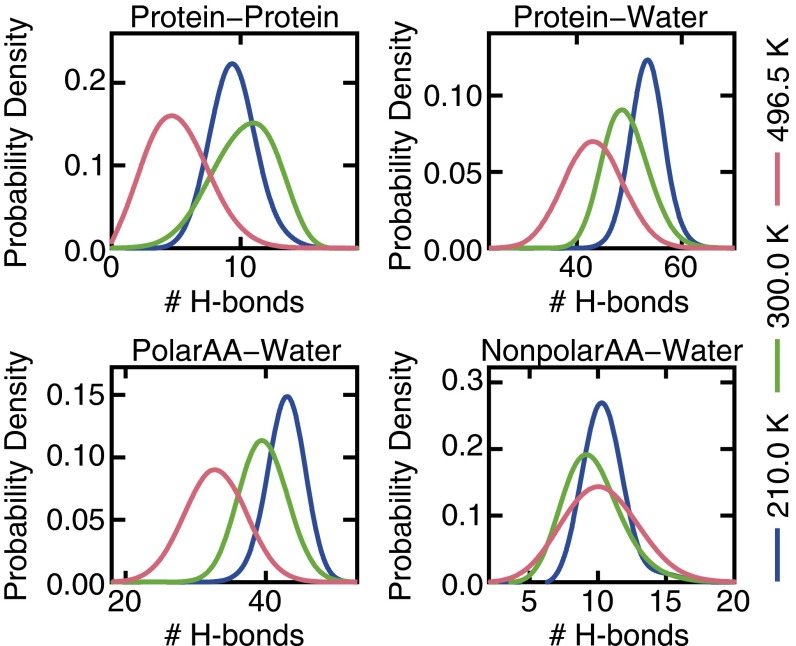
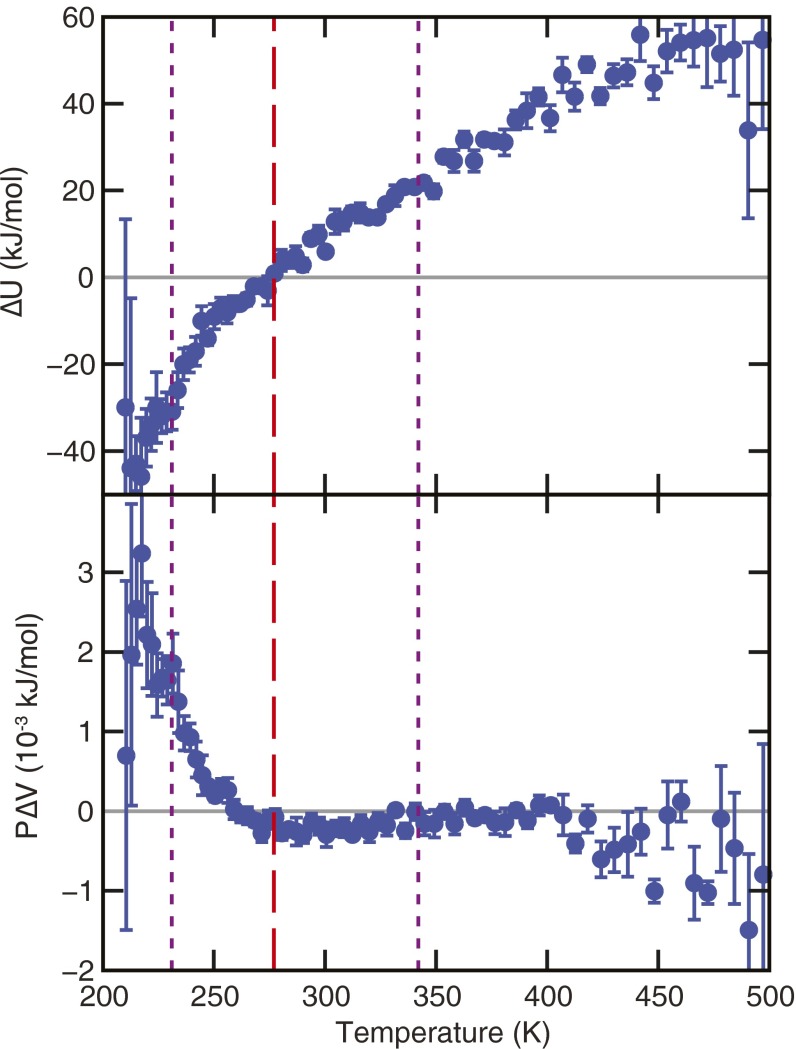
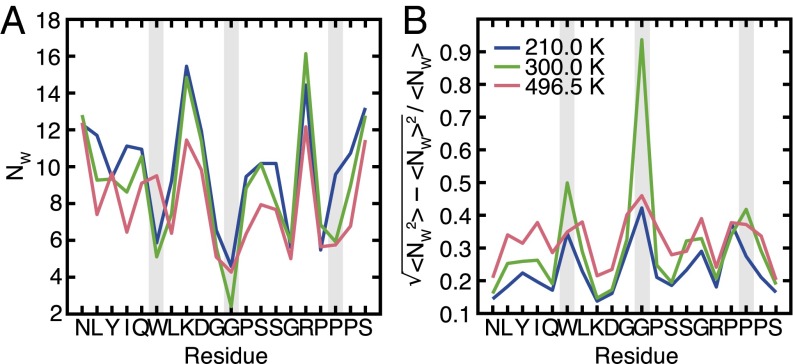
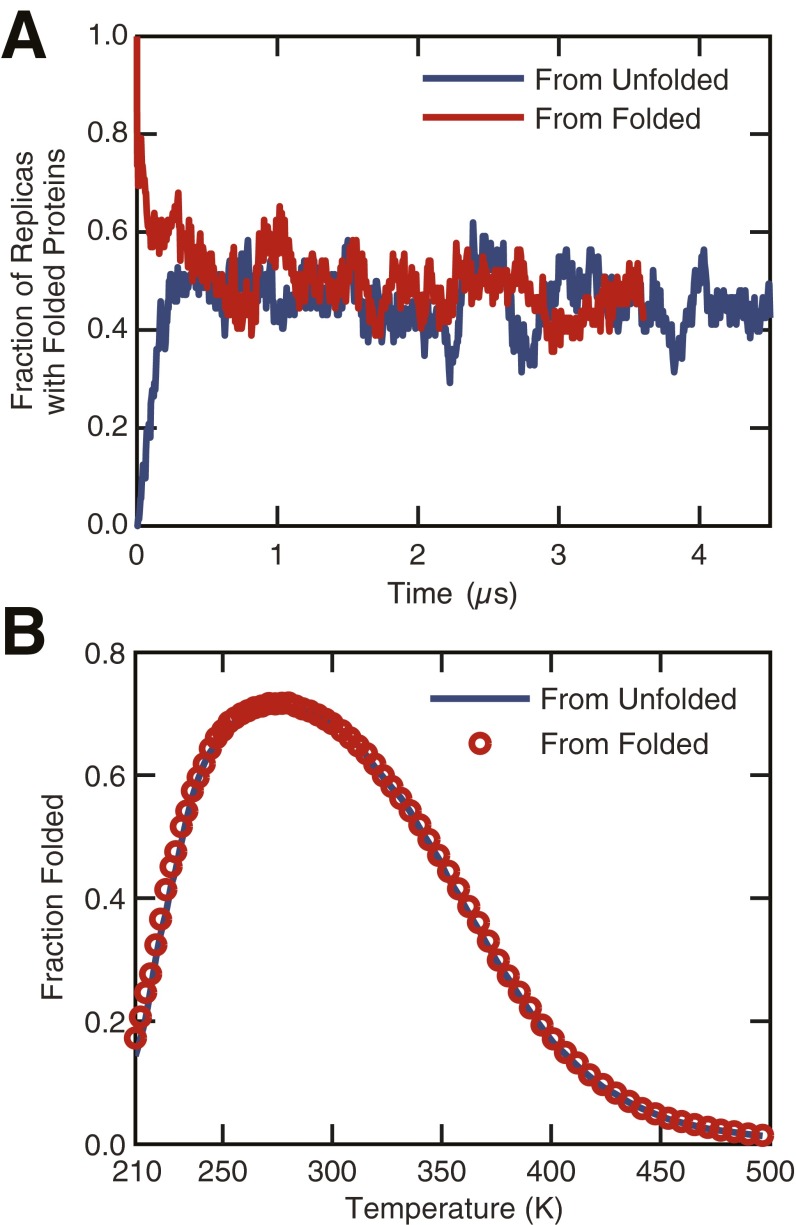
The functional native states of globular proteins become unstable at low temperatures, resulting in cold unfolding and impairment of normal biological function. Fundamental understanding of this phenomenon is essential to rationalizing the evolution of freeze-tolerant organisms and developing improved strategies for long-term preservation of biological materials. We present fully atomistic simulations of cold denaturation of an α-helical protein, the widely studied Trp-cage miniprotein. In contrast to the significant destabilization of the folded structure at high temperatures, Trp-cage cold denatures at 210 K into a compact, partially folded state; major elements of the secondary structure, including the α-helix, are conserved, but the salt bridge between aspartic acid and arginine is lost. The stability of Trp-cage's α-helix at low temperatures suggests a possible evolutionary explanation for the prevalence of such structures in antifreeze peptides produced by cold-weather species, such as Arctic char. Although the 310-helix is observed at cold conditions, its position is shifted toward Trp-cage's C-terminus. This shift is accompanied by intrusion of water into Trp-cage's interior and the hydration of buried hydrophobic residues. However, our calculations also show that the dominant contribution to the favorable energetics of low-temperature unfolding of Trp-cage comes from the hydration of hydrophilic residues.
Keywords: Trp-cage miniprotein; cold denaturation; protein folding.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Tanford C. Protein denaturation. Adv Protein Chem. 1968;23:121–282. - PubMed
-
- Zhang J, Peng X, Jonas A, Jonas J. NMR study of the cold, heat, and pressure unfolding of ribonuclease A. Biochemistry. 1995;34(27):8631–8641. - PubMed
-
- Privalov PL. Stability of proteins: Small globular proteins. Adv Protein Chem. 1979;33:167–241. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
