

The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine
- PMID: 27460824
- PMCID: PMC4962446
- DOI: 10.1186/s13073-016-0333-9
The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine
Abstract
Background: The diversity of clinical tumor profiling approaches (small panels to whole exomes with matched or unmatched germline analysis) may engender uncertainty about their benefits and liabilities, particularly in light of reported germline false positives in tumor-only profiling and use of global mutational and/or neoantigen data. The goal of this study was to determine the impact of genomic analysis strategies on error rates and data interpretation across contexts and ancestries.
Methods: We modeled common tumor profiling modalities-large (n = 300 genes), medium (n = 48 genes), and small (n = 15 genes) panels-using clinical whole exomes (WES) from 157 patients with lung or colon adenocarcinoma. We created a tumor-only analysis algorithm to assess germline false positive rates, the impact of patient ancestry on tumor-only results, and neoantigen detection.
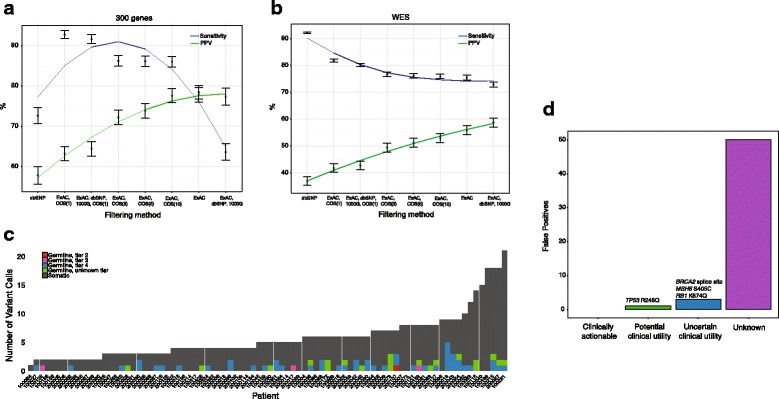
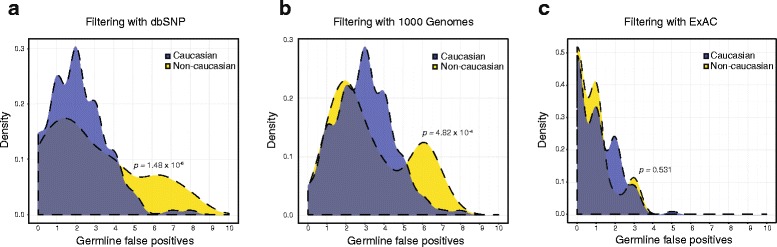
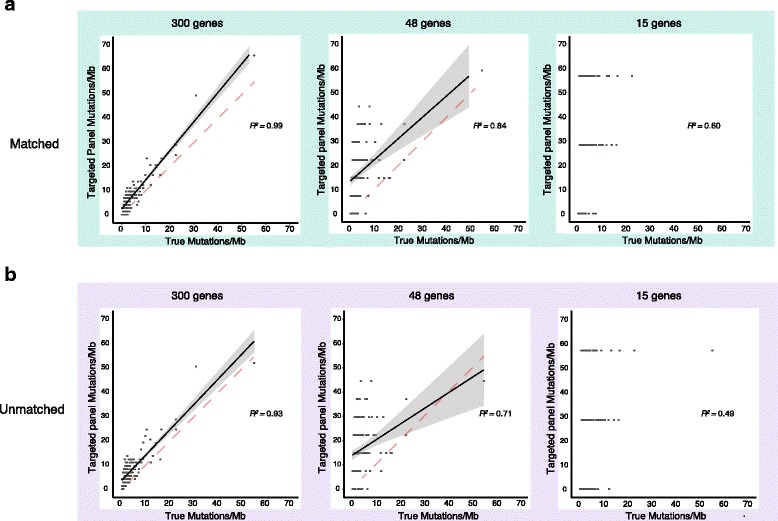
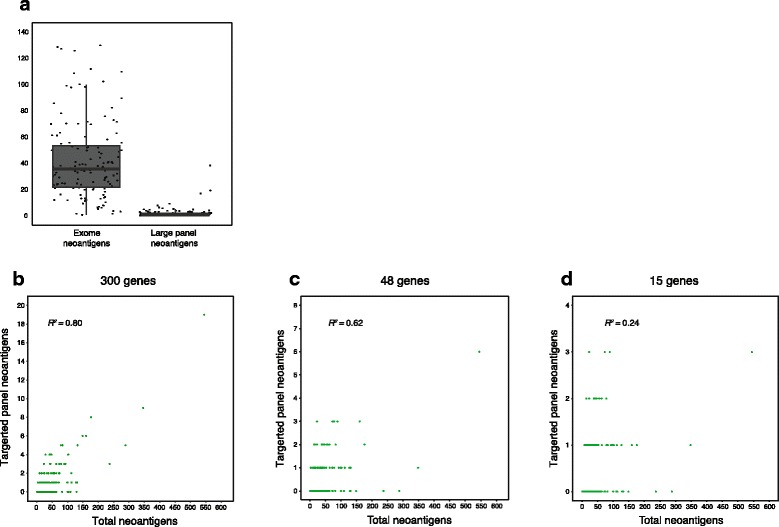
Results: After optimizing a germline filtering strategy, the germline false positive rate with tumor-only large panel sequencing was 14 % (144/1012 variants). For patients whose tumor-only results underwent molecular pathologist review (n = 91), 50/54 (93 %) false positives were correctly interpreted as uncertain variants. Increased germline false positives were observed in tumor-only sequencing of non-European compared with European ancestry patients (p < 0.001; Fisher's exact) when basic germline filtering approaches were used; however, the ExAC database (60,706 germline exomes) mitigated this disparity (p = 0.53). Matched and unmatched large panel mutational load correlated with WES mutational load (r(2) = 0.99 and 0.93, respectively; p < 0.001). Neoantigen load also correlated (r(2) = 0.80; p < 0.001), though WES identified a broader spectrum of neoantigens. Small panels did not predict mutational or neoantigen load.
Conclusions: Large tumor-only targeted panels are sufficient for most somatic variant identification and mutational load prediction if paired with expanded germline analysis strategies and molecular pathologist review. Paired germline sequencing reduced overall false positive mutation calls and WES provided the most neoantigens. Without patient-matched germline data, large germline databases are needed to minimize false positive mutation calling and mitigate ethnic disparities.
Keywords: Disparities; Genomics; Immuno-oncology; Neoantigens; Panel testing; Precision medicine.
Figures
References
-
- Garraway LA. Genomics-driven oncology: framework for an emerging paradigm. J Clin Oncol. 2013 - PubMed
-
- Pritchard CC, Salipante SJ, Koehler K, Smith C, Scroggins S, Wood B, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16(1):56–67. doi: 10.1016/j.jmoldx.2013.08.004. - DOI - PMC - PubMed
-
- Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251–264. doi: 10.1016/j.jmoldx.2014.12.006. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
