

A comparative study of k-spectrum-based error correction methods for next-generation sequencing data analysis
- PMID: 27461106
- PMCID: PMC4965716
- DOI: 10.1186/s40246-016-0068-0
A comparative study of k-spectrum-based error correction methods for next-generation sequencing data analysis
Abstract
Background: Innumerable opportunities for new genomic research have been stimulated by advancement in high-throughput next-generation sequencing (NGS). However, the pitfall of NGS data abundance is the complication of distinction between true biological variants and sequence error alterations during downstream analysis. Many error correction methods have been developed to correct erroneous NGS reads before further analysis, but independent evaluation of the impact of such dataset features as read length, genome size, and coverage depth on their performance is lacking. This comparative study aims to investigate the strength and weakness as well as limitations of some newest k-spectrum-based methods and to provide recommendations for users in selecting suitable methods with respect to specific NGS datasets.
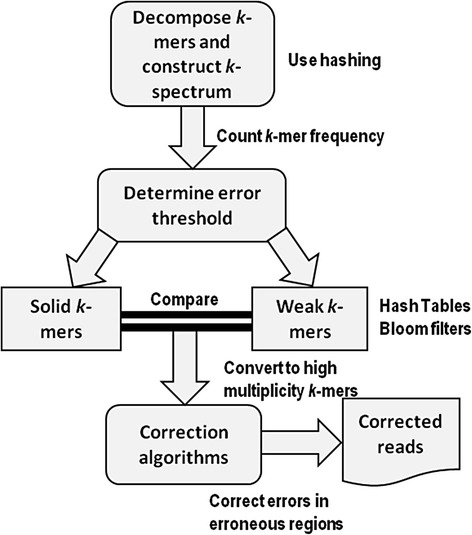
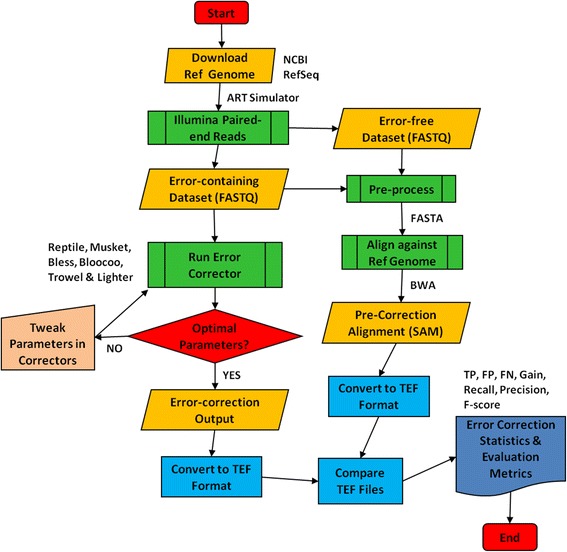
Methods: Six k-spectrum-based methods, i.e., Reptile, Musket, Bless, Bloocoo, Lighter, and Trowel, were compared using six simulated sets of paired-end Illumina sequencing data. These NGS datasets varied in coverage depth (10× to 120×), read length (36 to 100 bp), and genome size (4.6 to 143 MB). Error Correction Evaluation Toolkit (ECET) was employed to derive a suite of metrics (i.e., true positives, false positive, false negative, recall, precision, gain, and F-score) for assessing the correction quality of each method.
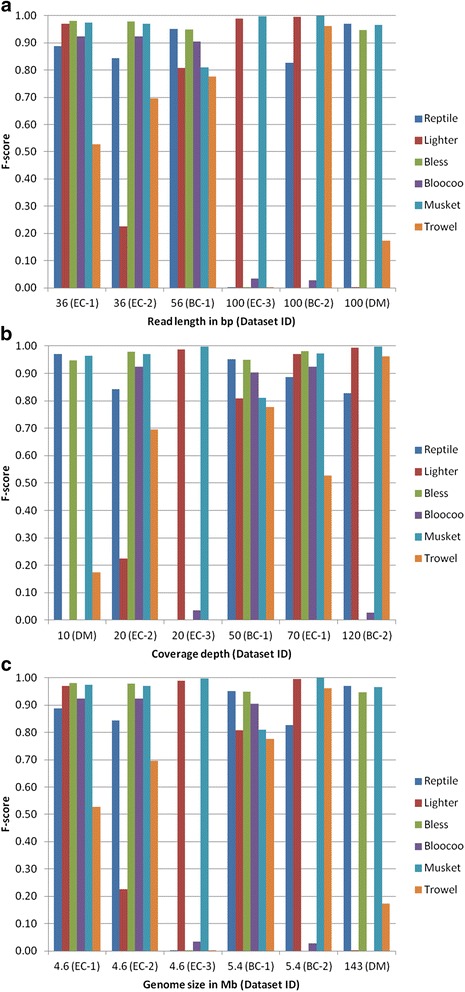
Results: Results from computational experiments indicate that Musket had the best overall performance across the spectra of examined variants reflected in the six datasets. The lowest accuracy of Musket (F-score = 0.81) occurred to a dataset with a medium read length (56 bp), a medium coverage (50×), and a small-sized genome (5.4 MB). The other five methods underperformed (F-score < 0.80) and/or failed to process one or more datasets.
Conclusions: This study demonstrates that various factors such as coverage depth, read length, and genome size may influence performance of individual k-spectrum-based error correction methods. Thus, efforts have to be paid in choosing appropriate methods for error correction of specific NGS datasets. Based on our comparative study, we recommend Musket as the top choice because of its consistently superior performance across all six testing datasets. Further extensive studies are warranted to assess these methods using experimental datasets generated by NGS platforms (e.g., 454, SOLiD, and Ion Torrent) under more diversified parameter settings (k-mer values and edit distances) and to compare them against other non-k-spectrum-based classes of error correction methods.
Keywords: Bloom filter; Error correction; Next-generation sequencing (NGS); Sequence analysis; k-mer; k-spectrum.
Figures
Similar articles
-
Athena: Automated Tuning of k-mer based Genomic Error Correction Algorithms using Language Models.Sci Rep. 2019 Nov 6;9(1):16157. doi: 10.1038/s41598-019-52196-4. Sci Rep. 2019. PMID: 31695060 Free PMC article.
-
QuorUM: An Error Corrector for Illumina Reads.PLoS One. 2015 Jun 17;10(6):e0130821. doi: 10.1371/journal.pone.0130821. eCollection 2015. PLoS One. 2015. PMID: 26083032 Free PMC article.
-
Trowel: a fast and accurate error correction module for Illumina sequencing reads.Bioinformatics. 2014 Nov 15;30(22):3264-5. doi: 10.1093/bioinformatics/btu513. Epub 2014 Jul 29. Bioinformatics. 2014. PMID: 25075116
-
A comprehensive evaluation of long read error correction methods.BMC Genomics. 2020 Dec 21;21(Suppl 6):889. doi: 10.1186/s12864-020-07227-0. BMC Genomics. 2020. PMID: 33349243 Free PMC article. Review.
-
Sequence assembly using next generation sequencing data--challenges and solutions.Sci China Life Sci. 2014 Nov;57(11):1140-8. doi: 10.1007/s11427-014-4752-9. Epub 2014 Oct 17. Sci China Life Sci. 2014. PMID: 25326069 Review.
Cited by
-
K-Mer Spectrum-Based Error Correction Algorithm for Next-Generation Sequencing Data.Comput Intell Neurosci. 2022 Jul 14;2022:8077664. doi: 10.1155/2022/8077664. eCollection 2022. Comput Intell Neurosci. 2022. PMID: 35875730 Free PMC article.
-
Lerna: transformer architectures for configuring error correction tools for short- and long-read genome sequencing.BMC Bioinformatics. 2022 Jan 6;23(1):25. doi: 10.1186/s12859-021-04547-0. BMC Bioinformatics. 2022. PMID: 34991450 Free PMC article.
-
Molecular characterization of an unauthorized genetically modified Bacillus subtilis production strain identified in a vitamin B2 feed additive.Food Chem. 2017 Sep 1;230:681-689. doi: 10.1016/j.foodchem.2017.03.042. Epub 2017 Mar 9. Food Chem. 2017. PMID: 28407967 Free PMC article.
-
HPTAS: An Alignment-Free Haplotype Phasing Algorithm Focused on Allele-Specific Studies Using Transcriptome Data.Int J Mol Sci. 2025 Jun 13;26(12):5700. doi: 10.3390/ijms26125700. Int J Mol Sci. 2025. PMID: 40565162 Free PMC article.
-
An overlooked phenomenon: complex interactions of potential error sources on the quality of bacterial de novo genome assemblies.BMC Genomics. 2024 Jan 9;25(1):45. doi: 10.1186/s12864-023-09910-4. BMC Genomics. 2024. PMID: 38195441 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
