

Characterization of VP1 sequence of Coxsackievirus A16 isolates by Bayesian evolutionary method
- PMID: 27464503
- PMCID: PMC4963925
- DOI: 10.1186/s12985-016-0578-3
Characterization of VP1 sequence of Coxsackievirus A16 isolates by Bayesian evolutionary method
Abstract
Background: Coxsackievirus A16 (CV-A16), a major etiopathologic cause of pediatric hand, foot, and mouth disease (HFMD) worldwide, has been reported to have caused several fatalities. Revealing the evolutionary and epidemiologic dynamics of CV-A16 across time and space is central to understanding its outbreak potential.
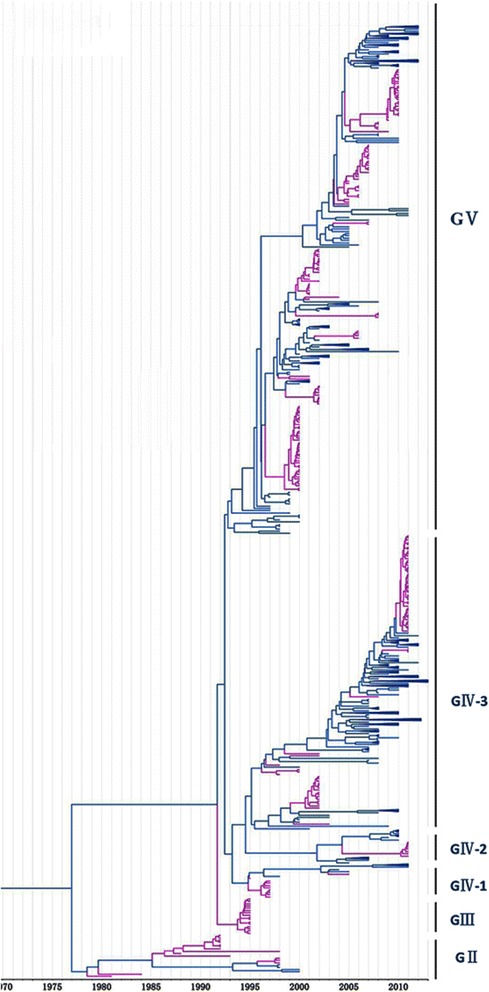
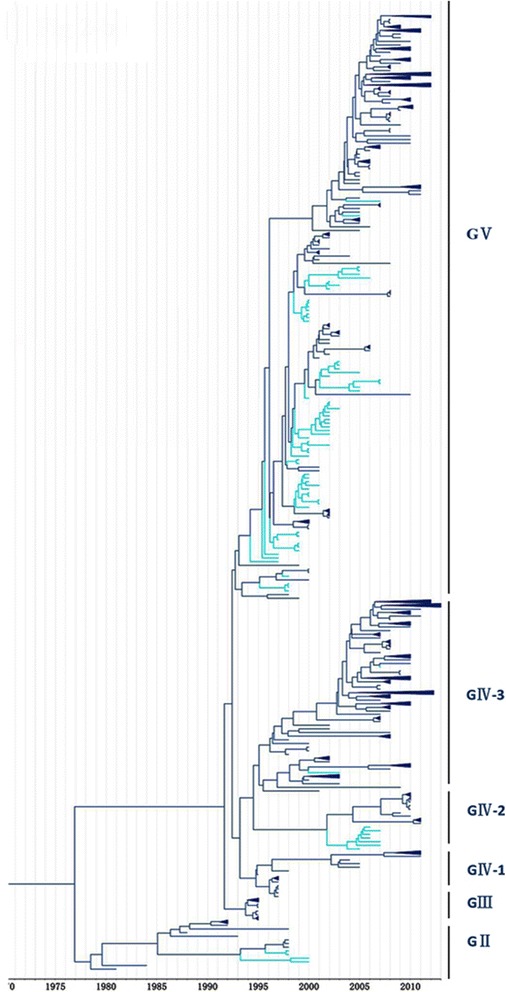
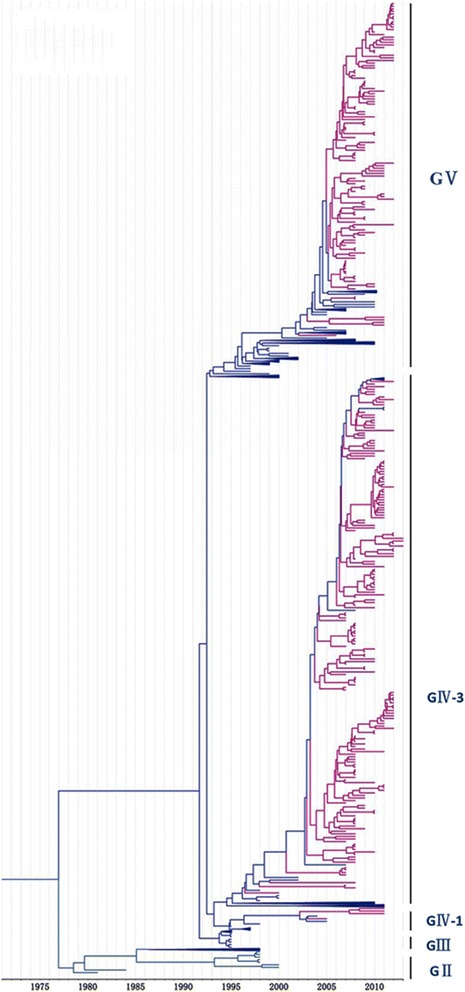
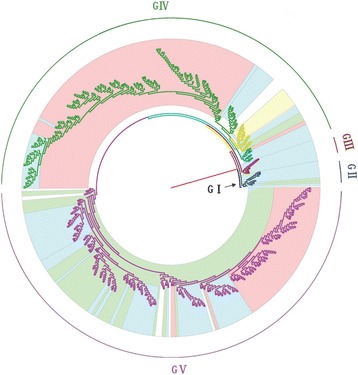
Methods: In this study, we isolated six CV-A16 strains in China's Jilin province and construct a maximum clade credibility (MCC) tree for CV-A16 VP1 gene by the Bayesian Markov Chain Monte Carlo method using 708 strains from GenBank with epidemiological information. The evolution characteristics of CV-A16 VP1 gene was also analysed dynamicly through Bayesian skyline plot.
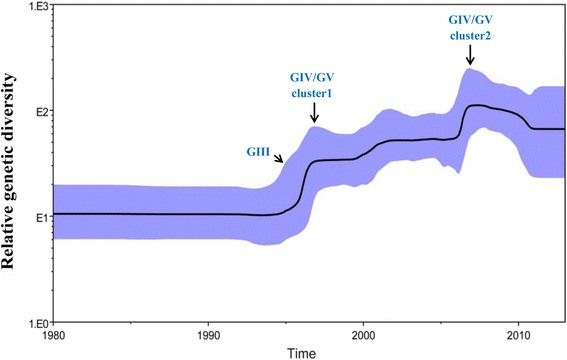
Results: All CV-A16 strains identified could be classified into five major genogroups, denoted by GI-GV. GIV and GV have co-circulated in China since 2007, and the CV-A16 epidemic strain isolated in the Jilin province, China, can be classified as GIV-3. The CV-A16 genogroups circulating recently in China have the same ancestor since 2007. The genetic diversity of the CV-A16 VP1 gene shows a continuous increase since the mid-1990s, with sharp increases in genetic diversity in 1997 and 2007 and reached peak in 2007. Very low genetic diversity existed after 2010. The CV-A16 VP1 gene evolutionary rate was 6.656E-3 substitutions per site per year.
Conclusions: We predicted the dynamic phylogenetic trends, which indicate outbreak trends of CV-A16, and provide theoretical foundations for clinical prevention and treatment of HFMD which caused by a CV-A16.
Keywords: Bayesian method; Coxsackievirus A16; Genetic diversity; HFMD; Molecular evolution.
Figures

References
-
- Shekhar K, Lye MS, Norlijah O, Ong F, Looi LM, Khuzaiah R, Marzuki I, Hussein I, Wong SL, Mohan J, et al. Deaths in children during an outbreak of hand, foot and mouth disease in Peninsular Malaysia--clinical and pathological characteristics. Med J Malaysia. 2005;60:297–304. - PubMed
-
- Tao ZX, Li Y, Wang HY, Song LZ, Liu GF, Liu Y, Lin XJ, Feng L, Yang H, Fan QY, Xu AQ. Genotype distribution of enterovirus A species isolated in Shandong Province, China. Bing Du Xue Bao. 2009;25:410–4. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
