

Scalable metagenomics alignment research tool (SMART): a scalable, rapid, and complete search heuristic for the classification of metagenomic sequences from complex sequence populations
- PMID: 27465705
- PMCID: PMC4963998
- DOI: 10.1186/s12859-016-1159-6
Scalable metagenomics alignment research tool (SMART): a scalable, rapid, and complete search heuristic for the classification of metagenomic sequences from complex sequence populations
Abstract
Background: Next generation sequencing technology has enabled characterization of metagenomics through massively parallel genomic DNA sequencing. The complexity and diversity of environmental samples such as the human gut microflora, combined with the sustained exponential growth in sequencing capacity, has led to the challenge of identifying microbial organisms by DNA sequence. We sought to validate a Scalable Metagenomics Alignment Research Tool (SMART), a novel searching heuristic for shotgun metagenomics sequencing results.
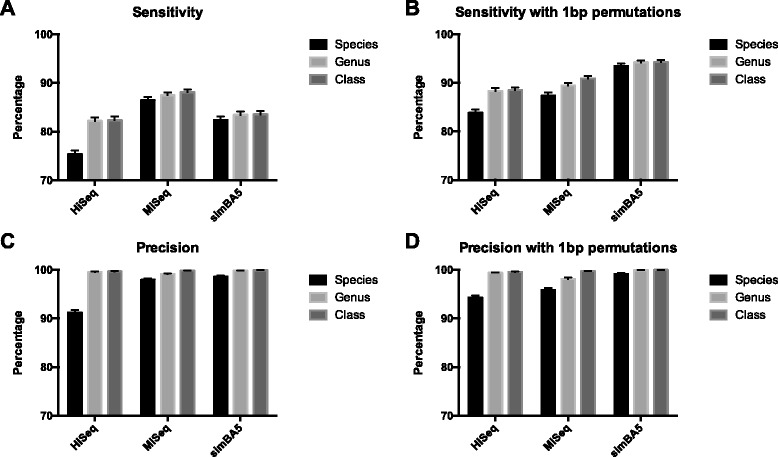
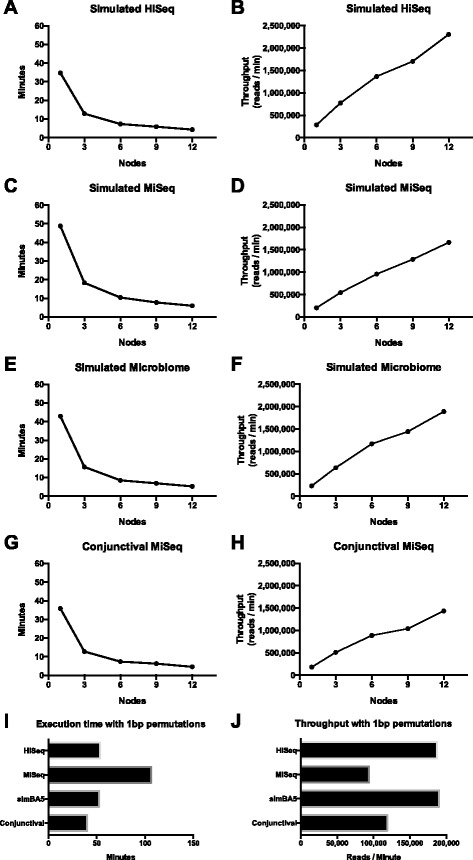
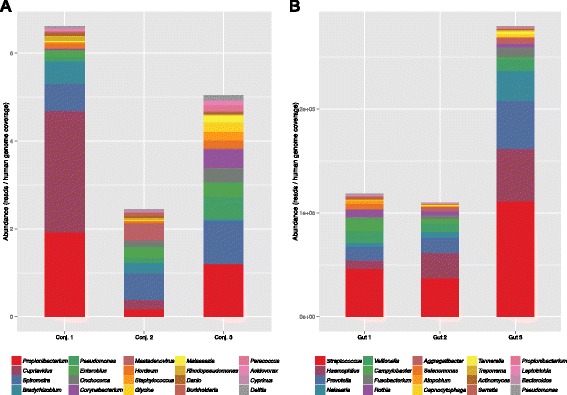
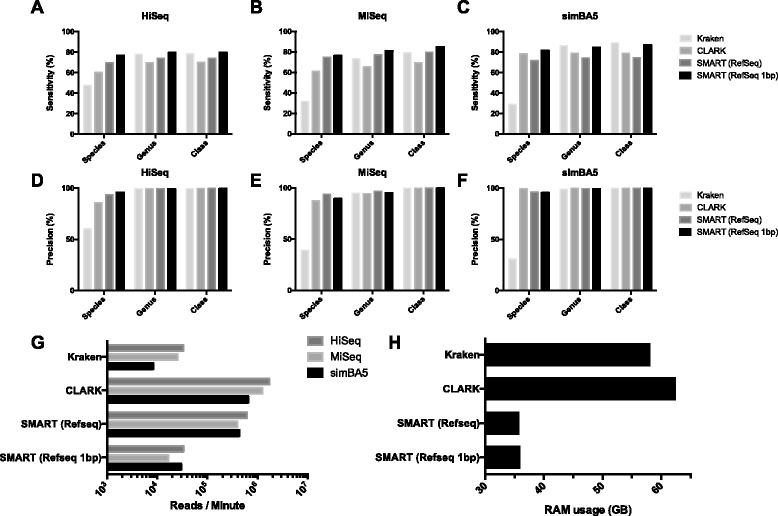
Results: After retrieving all genomic DNA sequences from the NCBI GenBank, over 1 × 10(11) base pairs of 3.3 × 10(6) sequences from 9.25 × 10(5) species were indexed using 4 base pair hashtable shards. A MapReduce searching strategy was used to distribute the search workload in a computing cluster environment. In addition, a one base pair permutation algorithm was used to account for single nucleotide polymorphisms and sequencing errors. Simulated datasets used to evaluate Kraken, a similar metagenomics classification tool, were used to measure and compare precision and accuracy. Finally using a same set of training sequences we compared Kraken, CLARK, and SMART within the same computing environment. Utilizing 12 computational nodes, we completed the classification of all datasets in under 10 min each using exact matching with an average throughput of over 1.95 × 10(6) reads classified per minute. With permutation matching, we achieved sensitivity greater than 83 % and precision greater than 94 % with simulated datasets at the species classification level. We demonstrated the application of this technique applied to conjunctival and gut microbiome metagenomics sequencing results. In our head to head comparison, SMART and CLARK had similar accuracy gains over Kraken at the species classification level, but SMART required approximately half the amount of RAM of CLARK.
Conclusions: SMART is the first scalable, efficient, and rapid metagenomics classification algorithm capable of matching against all the species and sequences present in the NCBI GenBank and allows for a single step classification of microorganisms as well as large plant, mammalian, or invertebrate genomes from which the metagenomic sample may have been derived.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
