

A Multi-scale Computational Platform to Mechanistically Assess the Effect of Genetic Variation on Drug Responses in Human Erythrocyte Metabolism
- PMID: 27467583
- PMCID: PMC4965186
- DOI: 10.1371/journal.pcbi.1005039
A Multi-scale Computational Platform to Mechanistically Assess the Effect of Genetic Variation on Drug Responses in Human Erythrocyte Metabolism
Abstract
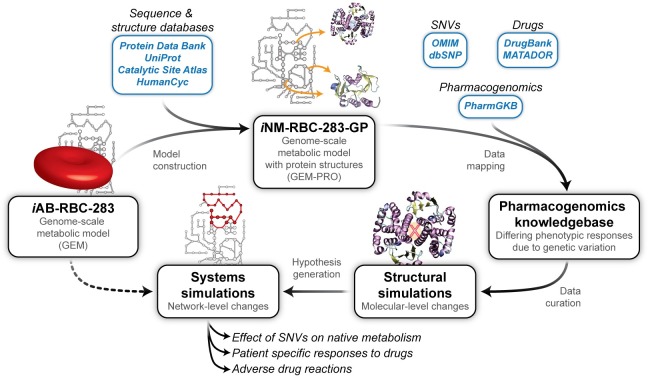
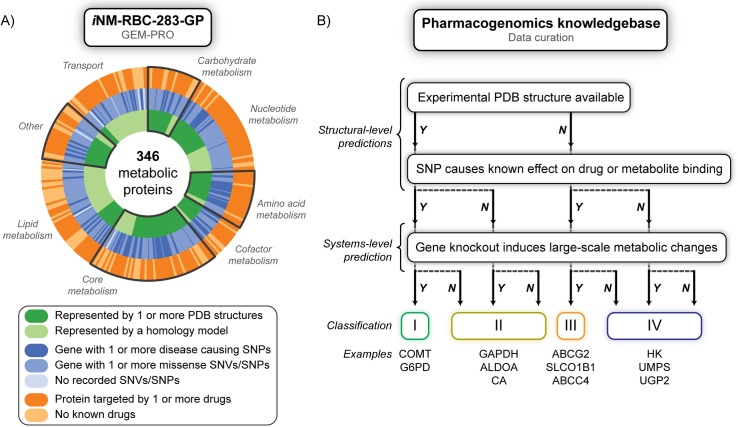
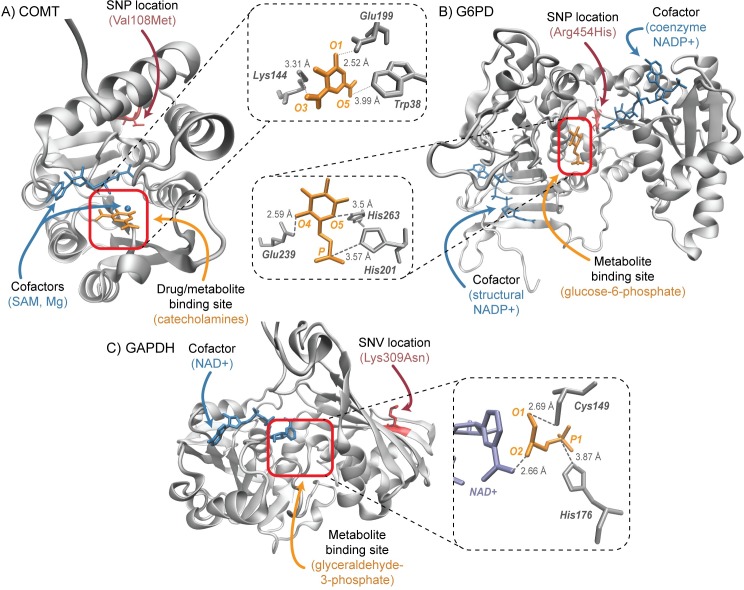
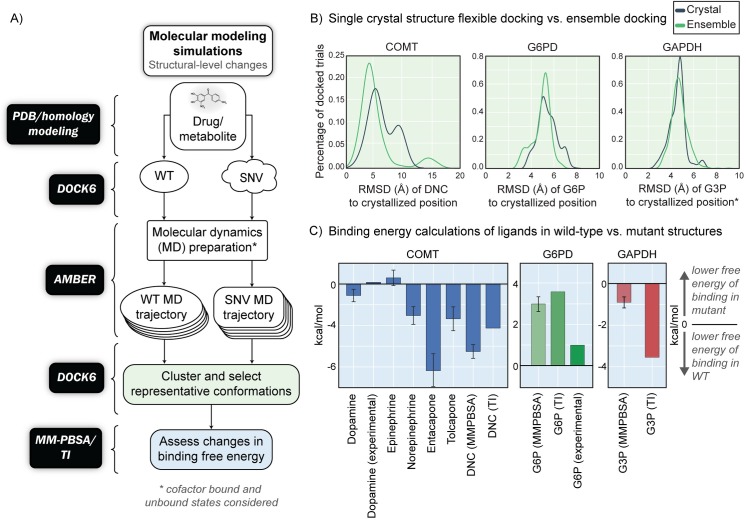
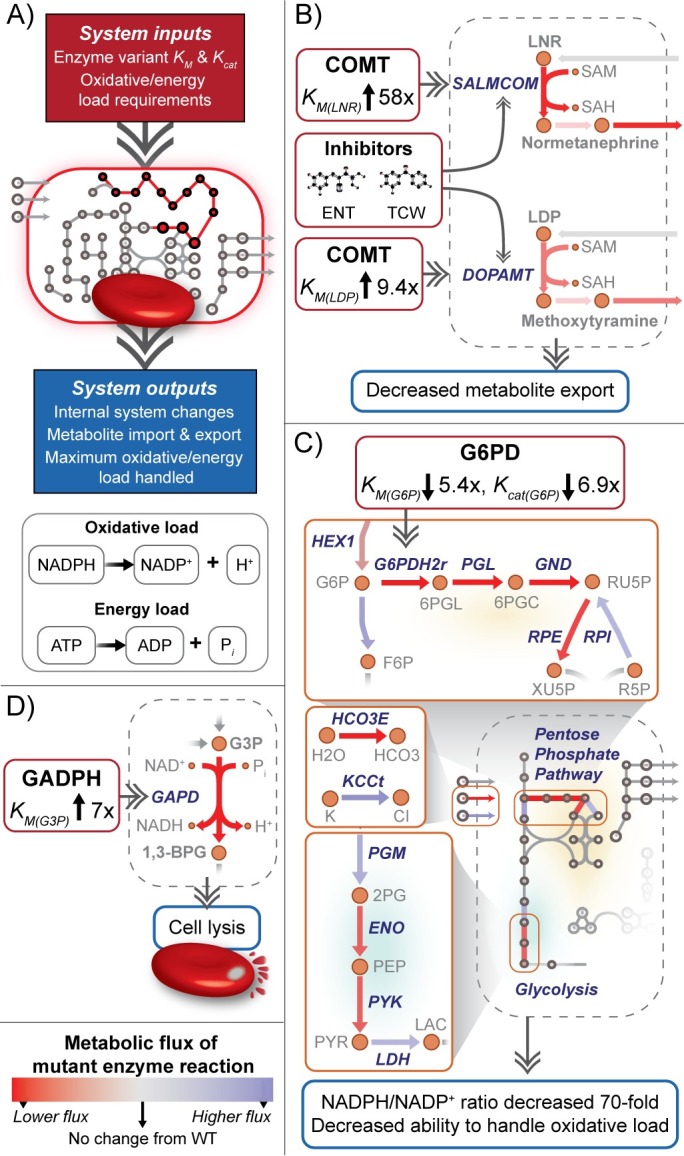
Progress in systems medicine brings promise to addressing patient heterogeneity and individualized therapies. Recently, genome-scale models of metabolism have been shown to provide insight into the mechanistic link between drug therapies and systems-level off-target effects while being expanded to explicitly include the three-dimensional structure of proteins. The integration of these molecular-level details, such as the physical, structural, and dynamical properties of proteins, notably expands the computational description of biochemical network-level properties and the possibility of understanding and predicting whole cell phenotypes. In this study, we present a multi-scale modeling framework that describes biological processes which range in scale from atomistic details to an entire metabolic network. Using this approach, we can understand how genetic variation, which impacts the structure and reactivity of a protein, influences both native and drug-induced metabolic states. As a proof-of-concept, we study three enzymes (catechol-O-methyltransferase, glucose-6-phosphate dehydrogenase, and glyceraldehyde-3-phosphate dehydrogenase) and their respective genetic variants which have clinically relevant associations. Using all-atom molecular dynamic simulations enables the sampling of long timescale conformational dynamics of the proteins (and their mutant variants) in complex with their respective native metabolites or drug molecules. We find that changes in a protein's structure due to a mutation influences protein binding affinity to metabolites and/or drug molecules, and inflicts large-scale changes in metabolism.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
