

Synthetic Applications of Proton-Coupled Electron Transfer
- PMID: 27472068
- PMCID: PMC5102158
- DOI: 10.1021/acs.accounts.6b00272
Synthetic Applications of Proton-Coupled Electron Transfer
Abstract
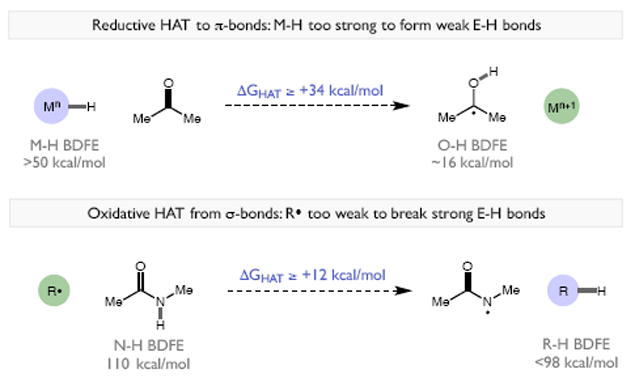
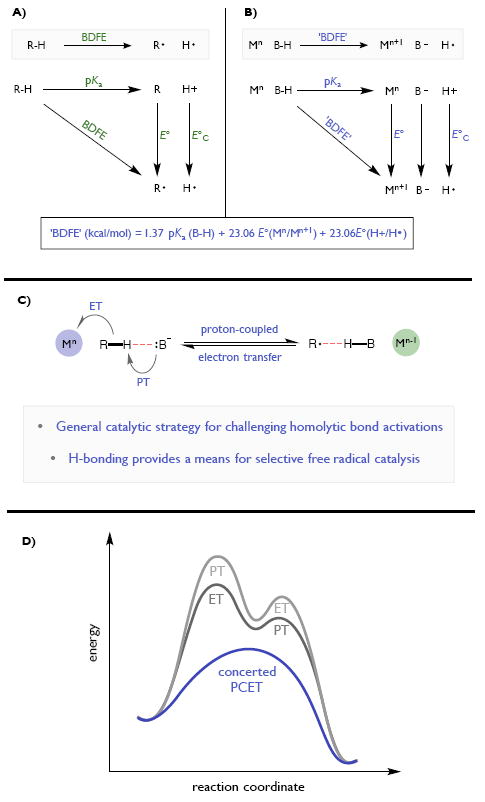
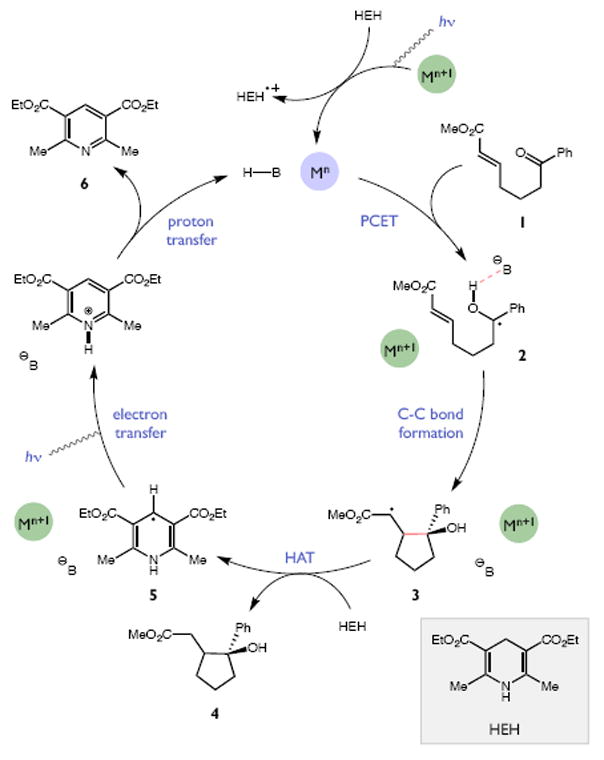
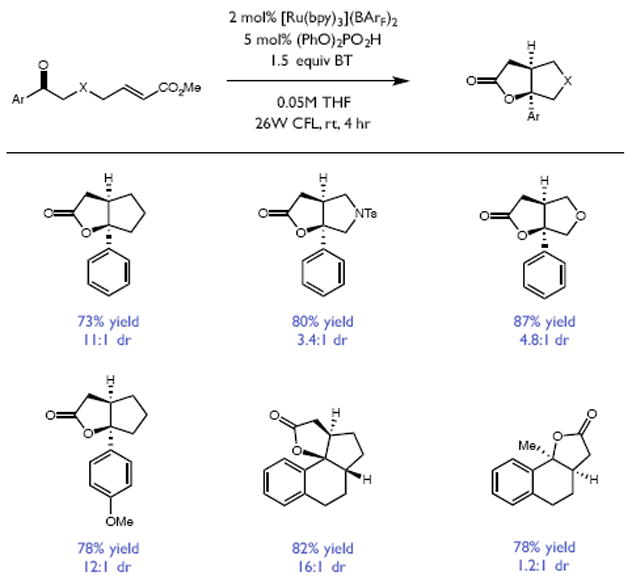
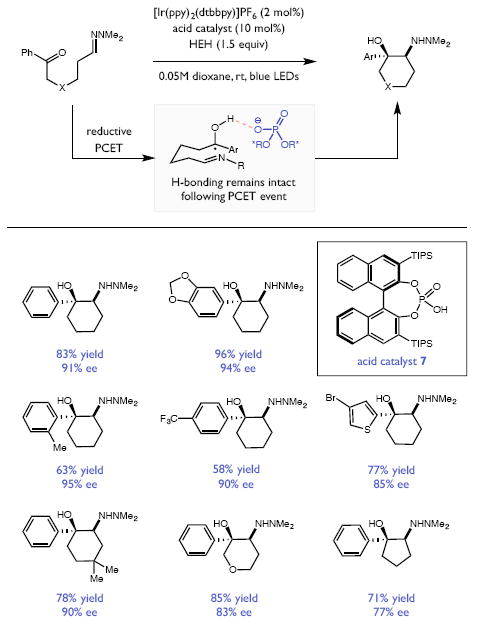
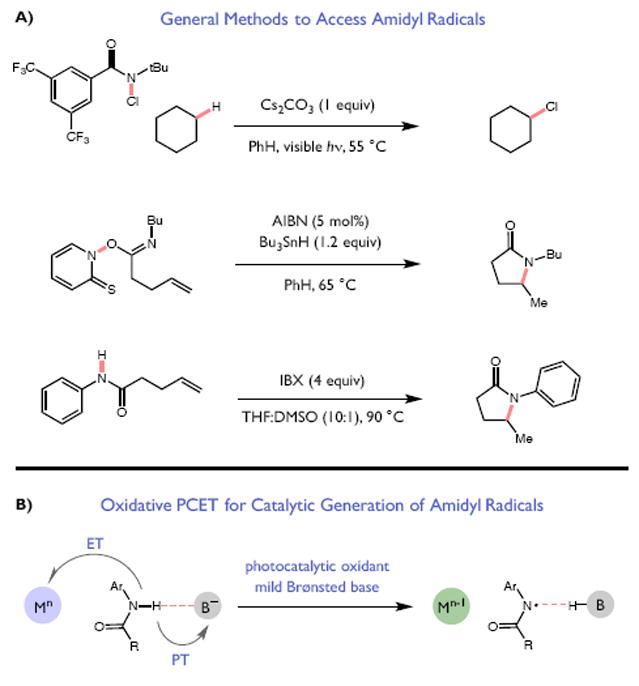
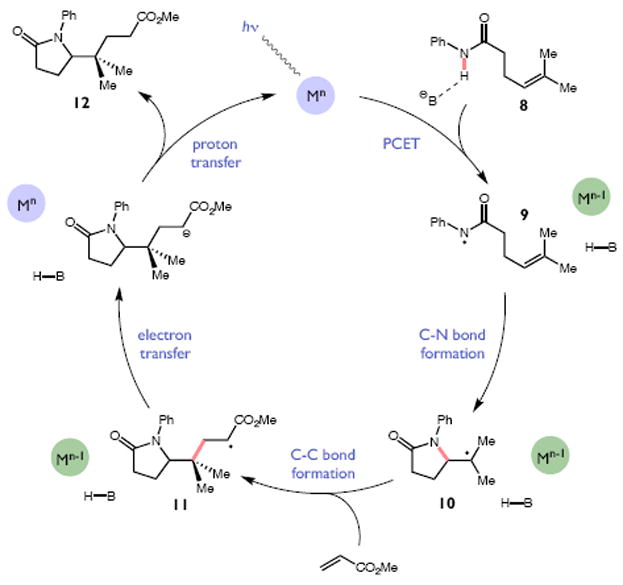
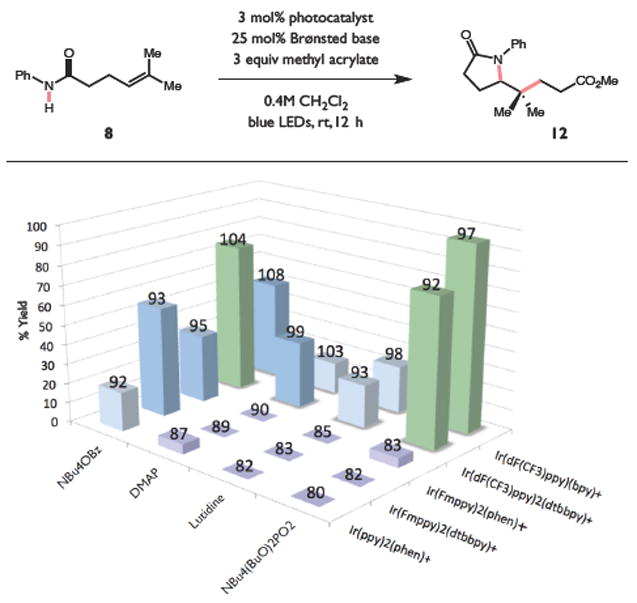
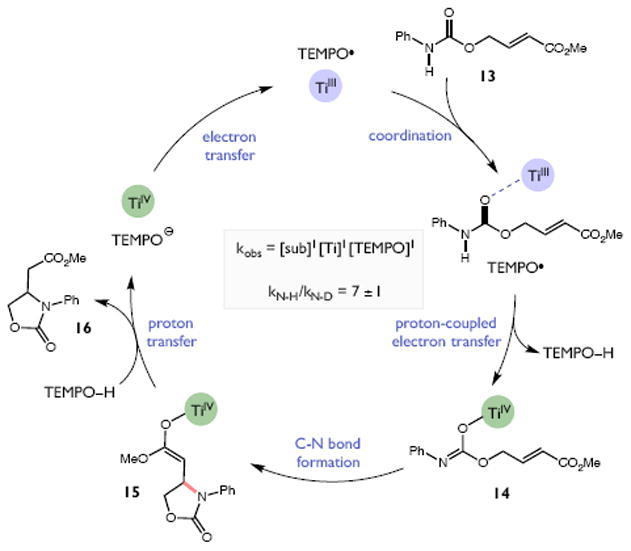
Redox events in which an electron and proton are exchanged in a concerted elementary step are commonly referred to as proton-coupled electron transfers (PCETs). PCETs are known to operate in numerous important biological redox processes, as well as recent inorganic technologies for small molecule activation. These studies suggest that PCET catalysis might also function as a general mode of substrate activation in organic synthesis. Over the past three years, our group has worked to advance this hypothesis and to demonstrate the synthetic utility of PCET through the development of novel catalytic radical chemistries. The central aim of these efforts has been to demonstrate the ability of PCET to homolytically activate a wide variety of common organic functional groups that are energetically inaccessible using known molecular H atom transfer catalysts. To do so, we made use of a simple formalism first introduced by Mayer and co-workers that allowed us to predict the thermodynamic capacity of any oxidant/base or reductant/acid pair to formally add or remove H· from a given substrate. With this insight, we were able to rationally select catalyst combinations thermodynamically competent to homolyze the extraordinarily strong E-H σ-bonds found in many common protic functional groups (BDFEs > 100 kcal/mol) or to form unusually weak bonds to hydrogen via the reductive action of common organic π-systems (BDFEs < 35 kcal/mol). These ideas were reduced to practice through the development of new catalyst systems for reductive PCET activations of ketones and oxidative PCET activation of amide N-H bonds to directly furnish reactive ketyl and amidyl radicals, respectively. In both systems, the reaction outcomes were found to be successfully predicted using the effective bond strength formalism, suggesting that these simple thermochemical considerations can provide useful and actionable insights into PCET reaction design. The ability of PCET catalysis to control enantioselectivity in free radical processes has also been established. Specifically, multisite PCET requires the formation of a pre-equilibrium hydrogen bond between the substrate and a proton donor/acceptor prior to charge transfer. We recognized that these H-bond interfaces persist following the PCET event, resulting in the formation of noncovalent complexes of the nascent radical intermediates. When chiral proton donors/acceptors are employed, this association can provide a basis for asymmetric induction in subsequent bond-forming steps. We discuss our efforts to capitalize on this understanding via the development of a catalytic protocol for enantioselective aza-pinacol cyclizations. Lastly, we highlight an alternative PCET mechanism that exploits the ability of redox-active metals to homolytically weaken the bonds in coordinated ligands, enabling nominally strong bonds (BDFEs ∼ 100 kcal) to be abstracted by weak H atom acceptors with concomitant oxidation of the metal center. This "soft homolysis" mechanism enables the generation of metalated intermediates from protic substrates under completely neutral conditions. The first example of this form of catalysis is presented in the context of a catalytic C-N bond forming reaction jointly mediated by bulky titanocene complexes and the stable nitroxyl radical TEMPO.
Conflict of interest statement
The authors declare no competing financial interest
Figures
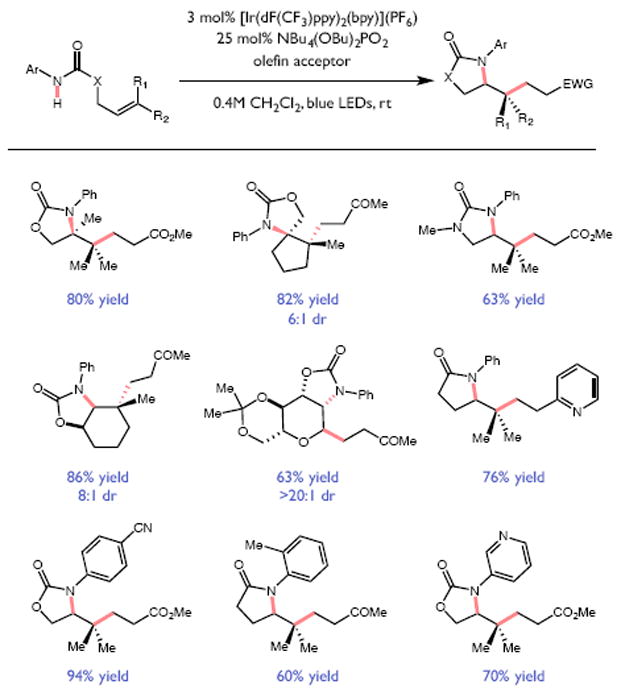
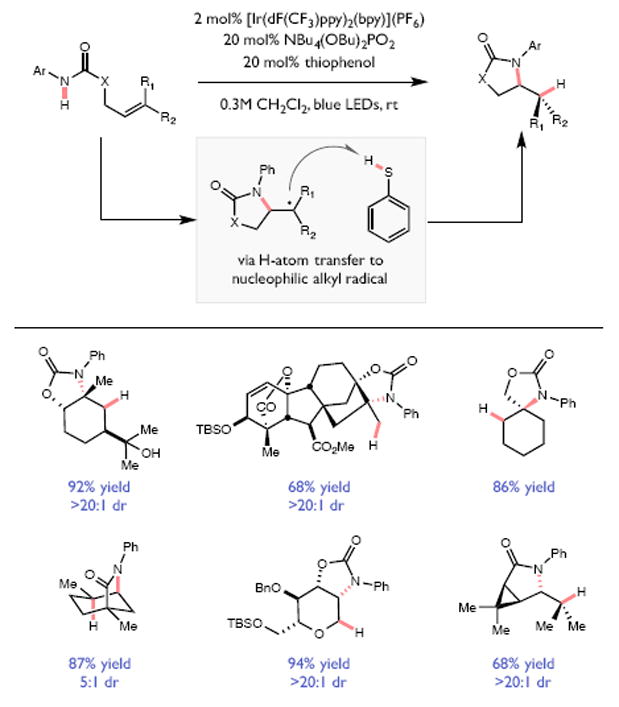
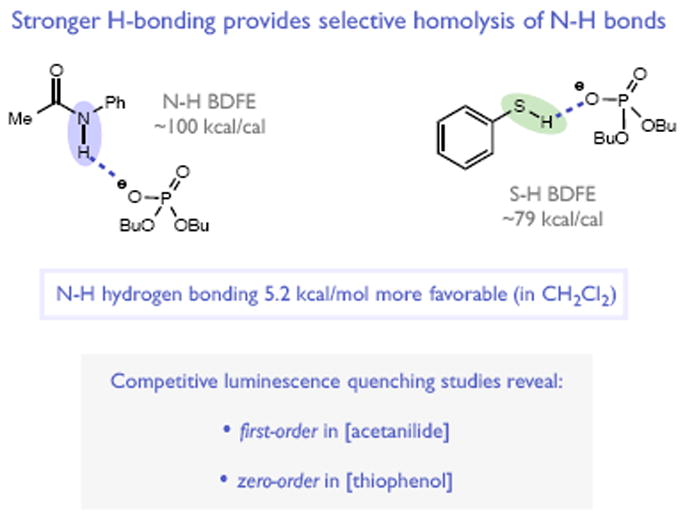
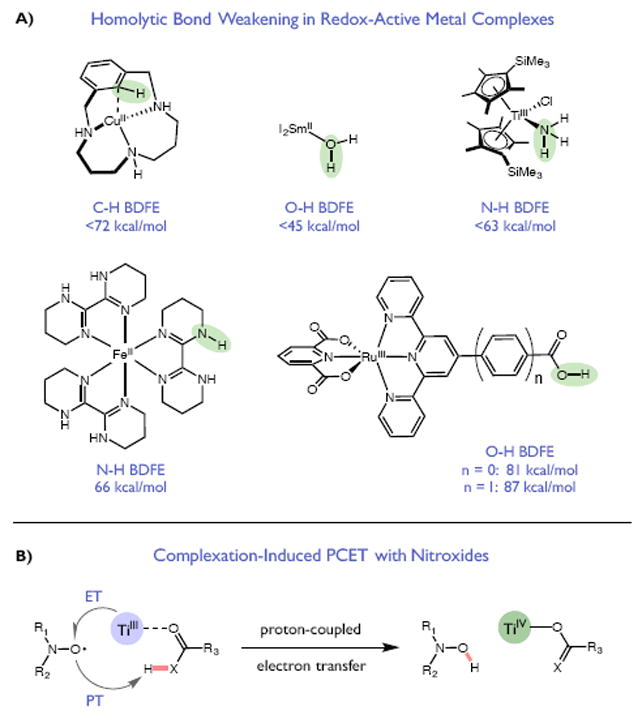
References
-
- Chen MS, White MC. Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science. 2010;327:566–571. - PubMed
-
- Liu W, Huang X, Cheng M-J, Nielsen RJ, Goddard WA, III, Groves JT. Oxidative Aliphatic C-H Fluorination with Fluoride Ion Catalyzed by a Manganese Porphyrin. Science. 2012;337:1322–1325. - PubMed
-
- Smith DM, Pulling ME, Norton JR. Tin-Free and Catalytic Radical Cyclizations. J Am Chem Soc. 2007;129:770–771. - PubMed
-
- Luo YR. Handbook of Bond Dissociation Free Energies in Organic Compounds. CRC Press; Boca Raton, FL: 2003.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
