

Deep learning for computational biology
- PMID: 27474269
- PMCID: PMC4965871
- DOI: 10.15252/msb.20156651
Deep learning for computational biology
Abstract
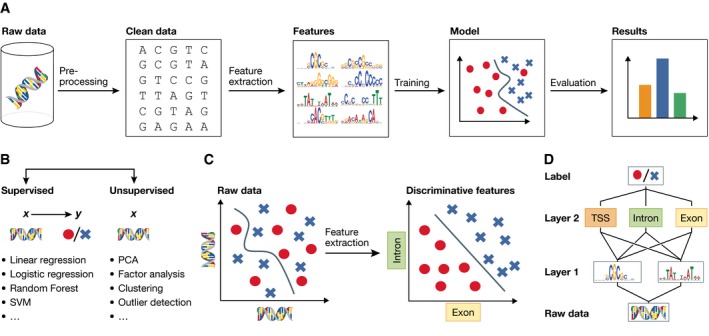
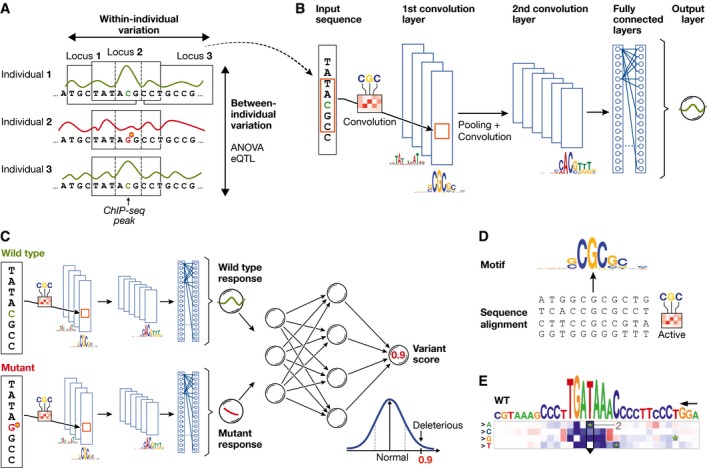
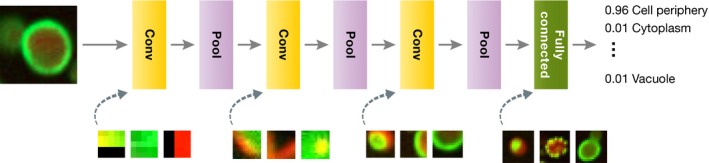
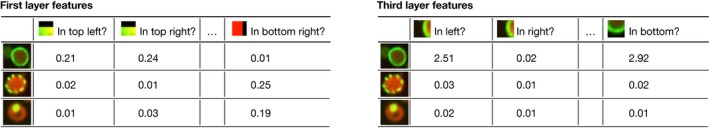
Technological advances in genomics and imaging have led to an explosion of molecular and cellular profiling data from large numbers of samples. This rapid increase in biological data dimension and acquisition rate is challenging conventional analysis strategies. Modern machine learning methods, such as deep learning, promise to leverage very large data sets for finding hidden structure within them, and for making accurate predictions. In this review, we discuss applications of this new breed of analysis approaches in regulatory genomics and cellular imaging. We provide background of what deep learning is, and the settings in which it can be successfully applied to derive biological insights. In addition to presenting specific applications and providing tips for practical use, we also highlight possible pitfalls and limitations to guide computational biologists when and how to make the most use of this new technology.
Keywords: cellular imaging; computational biology; deep learning; machine learning; regulatory genomics.
© 2016 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
References
-
- Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, Corrado GS, Davis A, Dean J, Devin M, Ghemawat S, Goodfellow I, Harp A, Irving G, Isard M, Jia Y, Josofowicz R, Kaiser L, Kudlur M, Levenberg J et al (2016) TensorFlow: large‐scale machine learning on heterogeneous distributed systems. arXiv:1603.04467
-
- Agathocleous M, Christodoulou G, Promponas V, Christodoulou C, Vassiliades V, Antoniou A (2010) Protein secondary structure prediction with bidirectional recurrent neural nets: can weight updating for each residue enhance performance? In Artificial Intelligence Applications and Innovations, Papadopoulos H, Andreou AS, Bramer M. (eds), Vol. 339, pp 128–137. Berlin Heidelberg: Springer;
-
- Alain G, Bengio Y, Rifai S (2012) Regularized auto‐encoders estimate local statistics. In Proc. CoRR, pp 1–17
-
- Alipanahi B, Delong A, Weirauch MT, Frey BJ (2015) Predicting the sequence specificities of DNA‐ and RNA‐binding proteins by deep learning. Nat Biotechnol 33: 831–838 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
