

Loss-of-Function Mutations in ELMO2 Cause Intraosseous Vascular Malformation by Impeding RAC1 Signaling
- PMID: 27476657
- PMCID: PMC4974086
- DOI: 10.1016/j.ajhg.2016.06.008
Loss-of-Function Mutations in ELMO2 Cause Intraosseous Vascular Malformation by Impeding RAC1 Signaling
Abstract
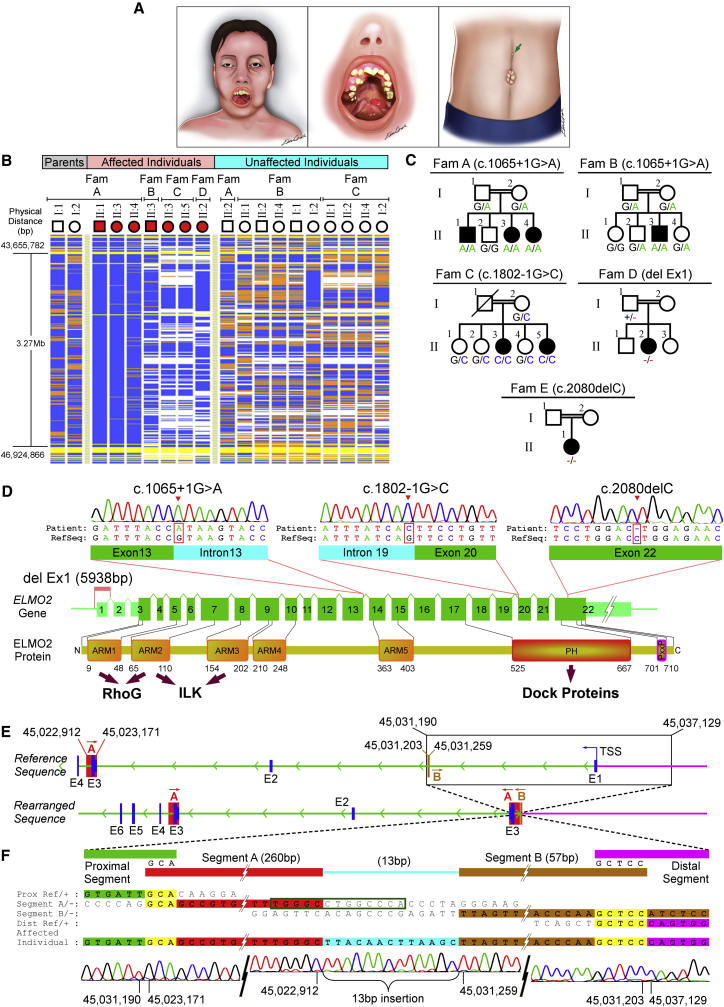
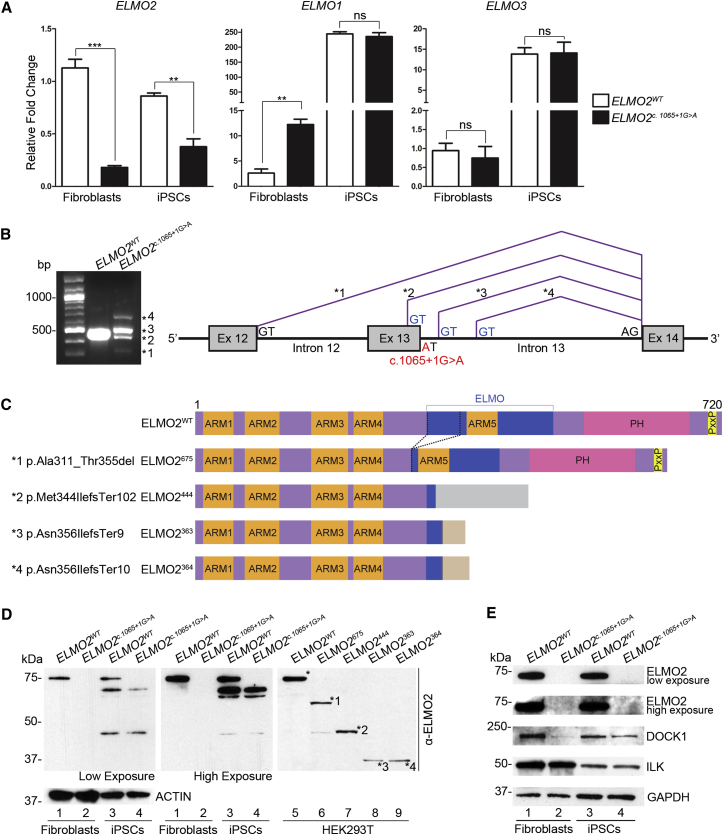
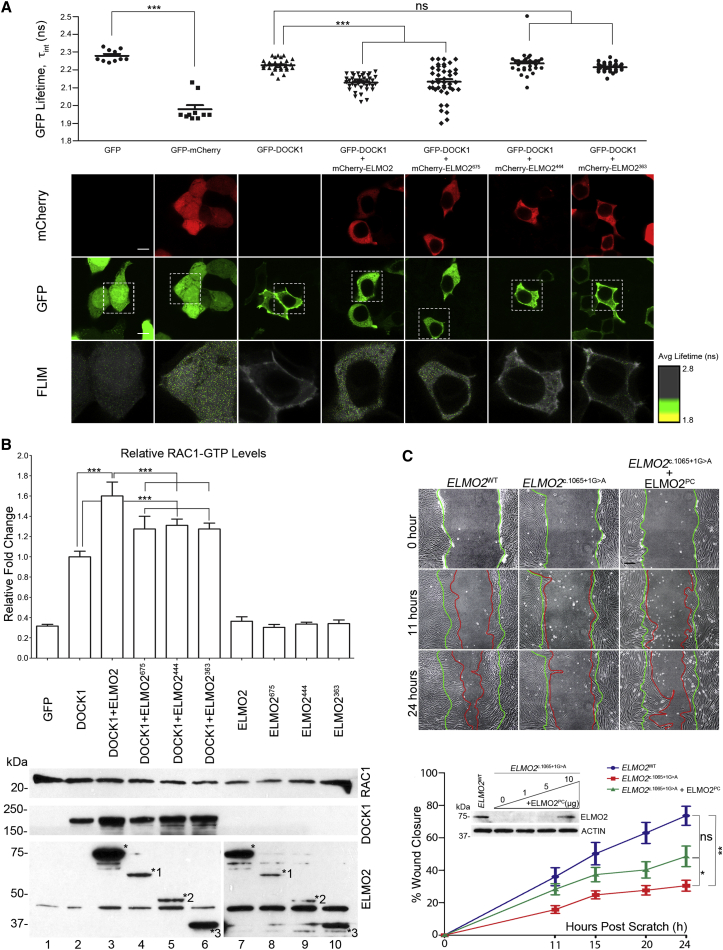
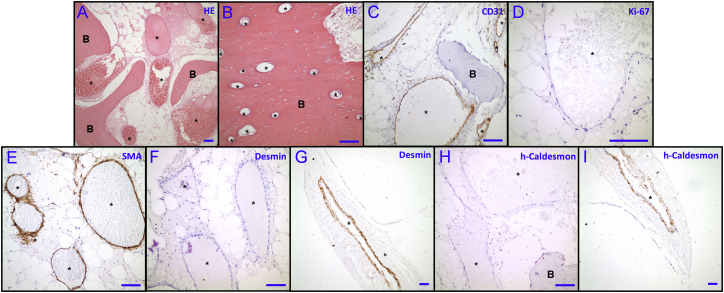
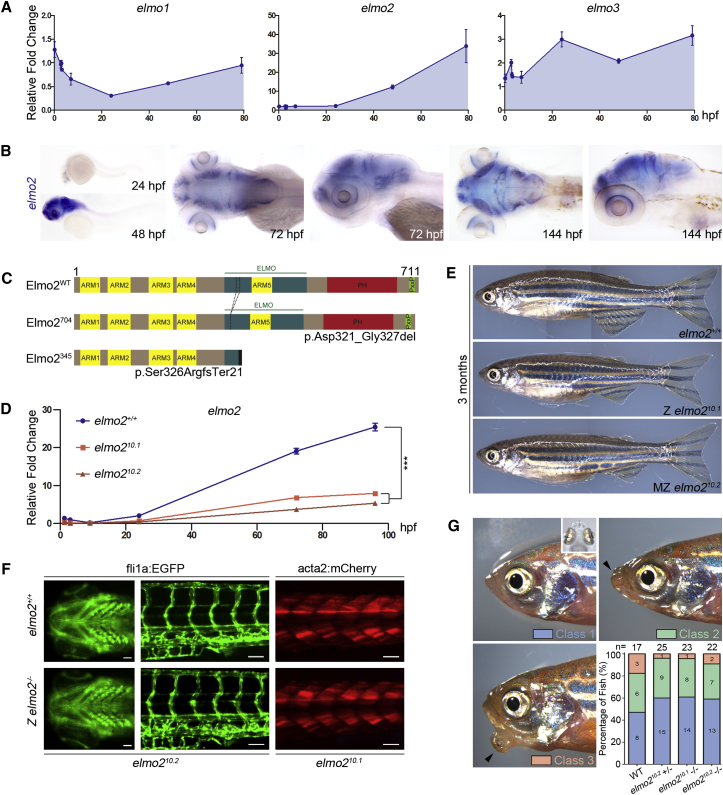
Vascular malformations are non-neoplastic expansions of blood vessels that arise due to errors during angiogenesis. They are a heterogeneous group of sporadic or inherited vascular disorders characterized by localized lesions of arteriovenous, capillary, or lymphatic origin. Vascular malformations that occur inside bone tissue are rare. Herein, we report loss-of-function mutations in ELMO2 (which translates extracellular signals into cellular movements) that are causative for autosomal-recessive intraosseous vascular malformation (VMOS) in five different families. Individuals with VMOS suffer from life-threatening progressive expansion of the jaw, craniofacial, and other intramembranous bones caused by malformed blood vessels that lack a mature vascular smooth muscle layer. Analysis of primary fibroblasts from an affected individual showed that absence of ELMO2 correlated with a significant downregulation of binding partner DOCK1, resulting in deficient RAC1-dependent cell migration. Unexpectedly, elmo2-knockout zebrafish appeared phenotypically normal, suggesting that there might be human-specific ELMO2 requirements in bone vasculature homeostasis or genetic compensation by related genes. Comparative phylogenetic analysis indicated that elmo2 originated upon the appearance of intramembranous bones and the jaw in ancestral vertebrates, implying that elmo2 might have been involved in the evolution of these novel traits. The present findings highlight the necessity of ELMO2 for maintaining vascular integrity, specifically in intramembranous bones.
Copyright © 2016 American Society of Human Genetics. All rights reserved.
Figures
Similar articles
-
ELMO2 is an essential regulator of carotid artery development.Nat Commun. 2025 Jun 2;16(1):5108. doi: 10.1038/s41467-025-60105-9. Nat Commun. 2025. PMID: 40456777 Free PMC article.
-
ELMO2 biallelic pathogenic variants in a patient with gingival hypertrophy and cherubism phenotype: Case report and molecular review.Am J Med Genet A. 2024 Oct;194(10):e63602. doi: 10.1002/ajmg.a.63602. Epub 2024 Mar 22. Am J Med Genet A. 2024. PMID: 38517102 Review.
-
Involvement of Tiam1, RhoG and ELMO2/ILK in Rac1-mediated phagocytosis in human trabecular meshwork cells.Exp Cell Res. 2016 Oct 1;347(2):301-11. doi: 10.1016/j.yexcr.2016.08.009. Epub 2016 Aug 15. Exp Cell Res. 2016. PMID: 27539661 Free PMC article.
-
Elmo2 Is a Regulator of Insulin-dependent Glut4 Membrane Translocation.J Biol Chem. 2016 Jul 29;291(31):16150-61. doi: 10.1074/jbc.M116.731521. Epub 2016 May 20. J Biol Chem. 2016. PMID: 27226625 Free PMC article.
-
Emerging importance of molecular pathogenesis of vascular malformations in clinical practice and classifications.Vasc Med. 2020 Aug;25(4):364-377. doi: 10.1177/1358863X20918941. Epub 2020 Jun 22. Vasc Med. 2020. PMID: 32568624 Review.
Cited by
-
FBXO22 deficiency defines a pleiotropic syndrome of growth restriction and multi-system anomalies associated with a unique epigenetic signature.Am J Hum Genet. 2025 May 1;112(5):1233-1246. doi: 10.1016/j.ajhg.2025.03.013. Epub 2025 Apr 10. Am J Hum Genet. 2025. PMID: 40215970
-
DOCK3-Associated Neurodevelopmental Disorder-Clinical Features and Molecular Basis.Genes (Basel). 2023 Oct 14;14(10):1940. doi: 10.3390/genes14101940. Genes (Basel). 2023. PMID: 37895289 Free PMC article. Review.
-
The DOCK protein family in vascular development and disease.Angiogenesis. 2021 Aug;24(3):417-433. doi: 10.1007/s10456-021-09768-8. Epub 2021 Feb 6. Angiogenesis. 2021. PMID: 33548004 Free PMC article. Review.
-
Mutations in MC4R facilitate the angiogenic activity in patients with orbital venous malformation.Exp Biol Med (Maywood). 2020 Jun;245(11):956-963. doi: 10.1177/1535370220919056. Epub 2020 May 3. Exp Biol Med (Maywood). 2020. PMID: 32363922 Free PMC article.
-
Structure of BAI1/ELMO2 complex reveals an action mechanism of adhesion GPCRs via ELMO family scaffolds.Nat Commun. 2019 Jan 3;10(1):51. doi: 10.1038/s41467-018-07938-9. Nat Commun. 2019. PMID: 30604775 Free PMC article.
References
-
- Conway E.M., Collen D., Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc. Res. 2001;49:507–521. - PubMed
-
- Mulliken J.B., Glowacki J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast. Reconstr. Surg. 1982;69:412–422. - PubMed
-
- Bruder E., Perez-Atayde A.R., Jundt G., Alomari A.I., Rischewski J., Fishman S.J., Mulliken J.B., Kozakewich H.P. Vascular lesions of bone in children, adolescents, and young adults. A clinicopathologic reappraisal and application of the ISSVA classification. Virchows Arch. 2009;454:161–179. - PubMed
-
- Wassef M., Blei F., Adams D., Alomari A., Baselga E., Berenstein A., Burrows P., Frieden I.J., Garzon M.C., Lopez-Gutierrez J.C., ISSVA Board and Scientific Committee Vascular anomalies classification: recommendations from the International Society for the Study of Vascular Anomalies. Pediatrics. 2015;136:e203–e214. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
