

High-throughput Phenotyping of Lung Cancer Somatic Mutations
- PMID: 27478040
- PMCID: PMC5003022
- DOI: 10.1016/j.ccell.2016.06.022
High-throughput Phenotyping of Lung Cancer Somatic Mutations
Erratum in
-
High-throughput Phenotyping of Lung Cancer Somatic Mutations.Cancer Cell. 2017 Dec 11;32(6):884. doi: 10.1016/j.ccell.2017.11.008. Cancer Cell. 2017. PMID: 29232558 No abstract available.
Abstract
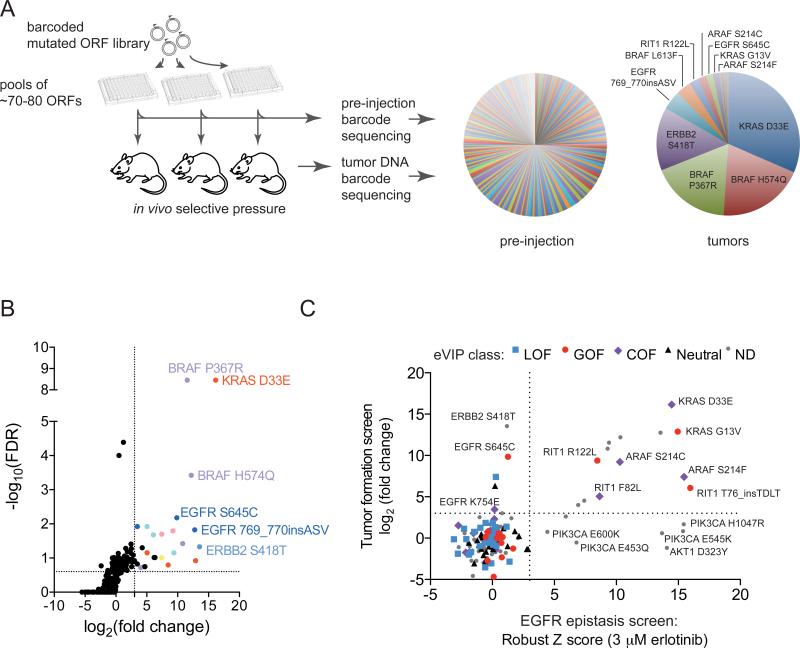
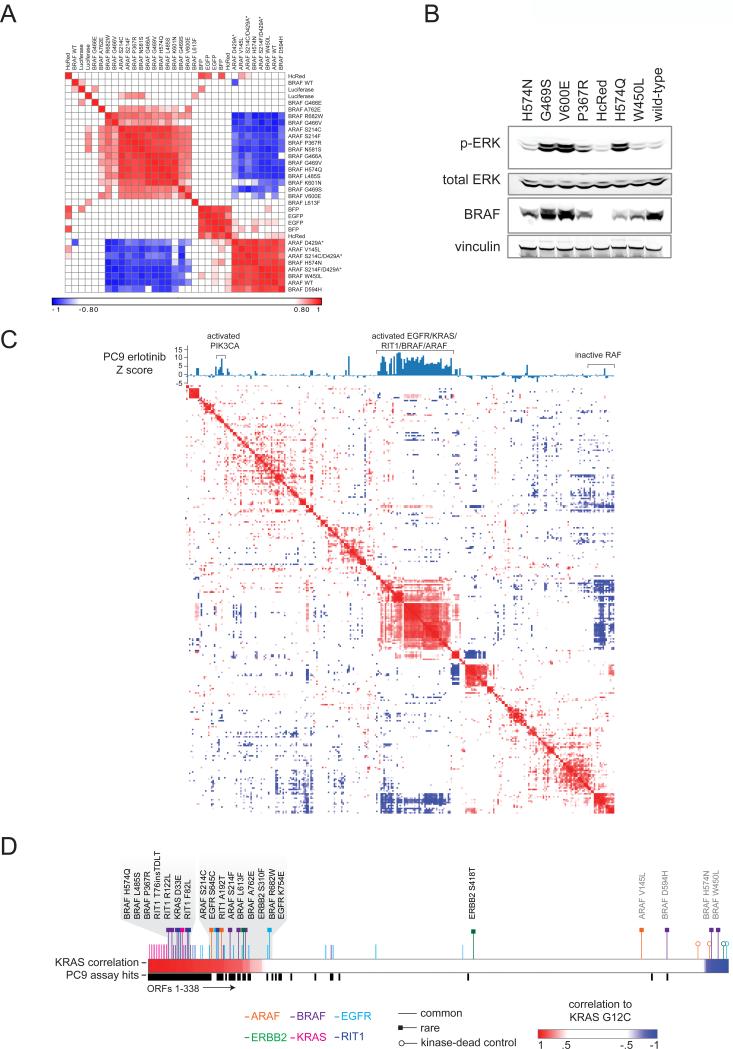
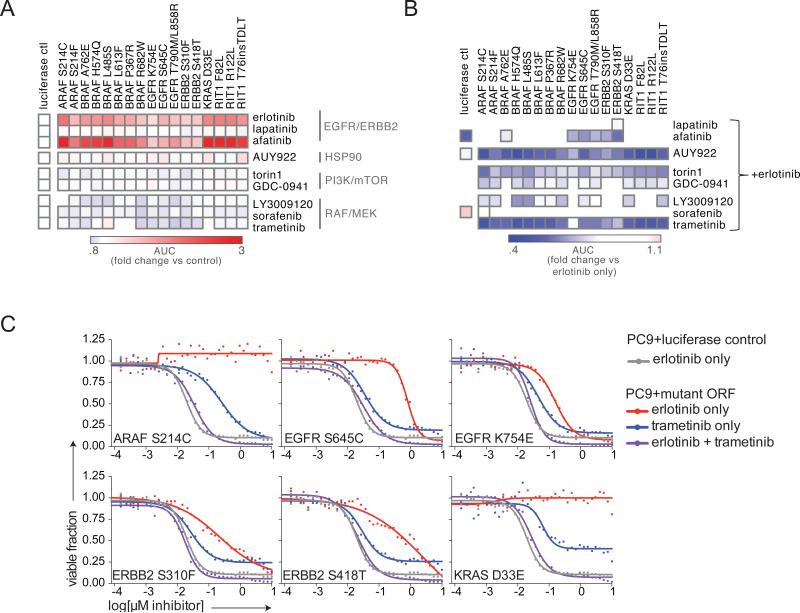
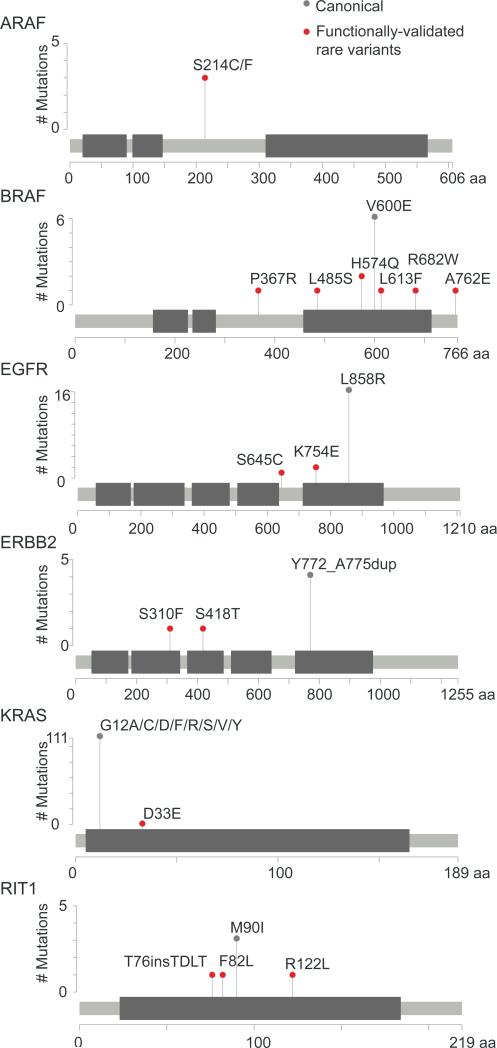
Recent genome sequencing efforts have identified millions of somatic mutations in cancer. However, the functional impact of most variants is poorly understood. Here we characterize 194 somatic mutations identified in primary lung adenocarcinomas. We present an expression-based variant-impact phenotyping (eVIP) method that uses gene expression changes to distinguish impactful from neutral somatic mutations. eVIP identified 69% of mutations analyzed as impactful and 31% as functionally neutral. A subset of the impactful mutations induces xenograft tumor formation in mice and/or confers resistance to cellular EGFR inhibition. Among these impactful variants are rare somatic, clinically actionable variants including EGFR S645C, ARAF S214C and S214F, ERBB2 S418T, and multiple BRAF variants, demonstrating that rare mutations can be functionally important in cancer.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
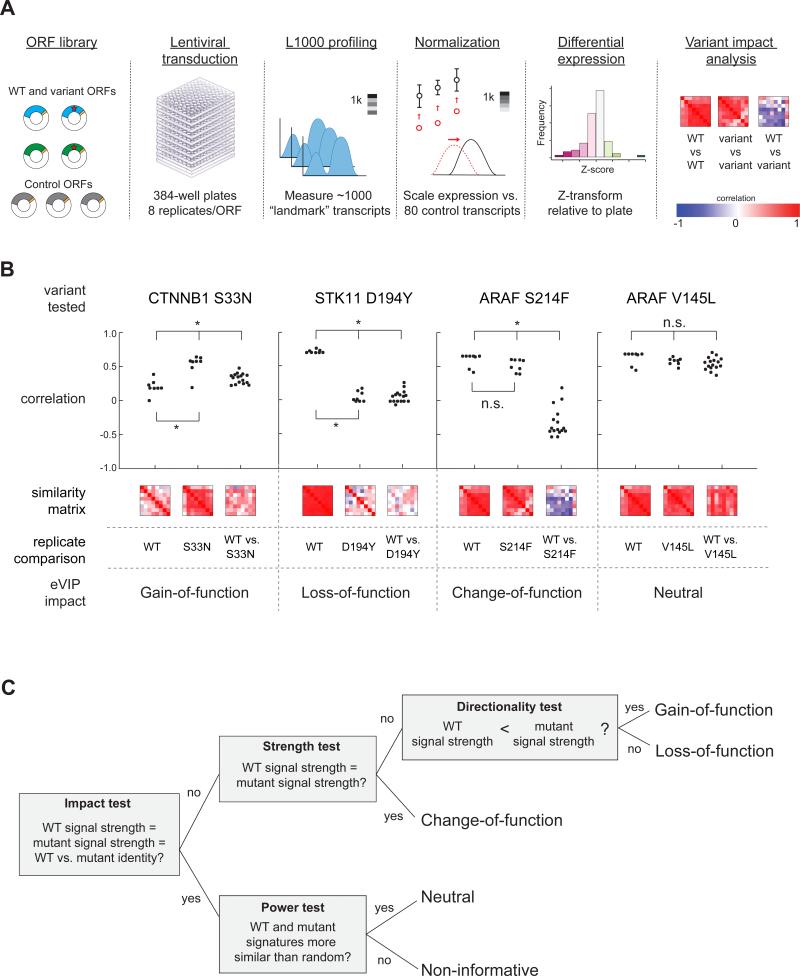
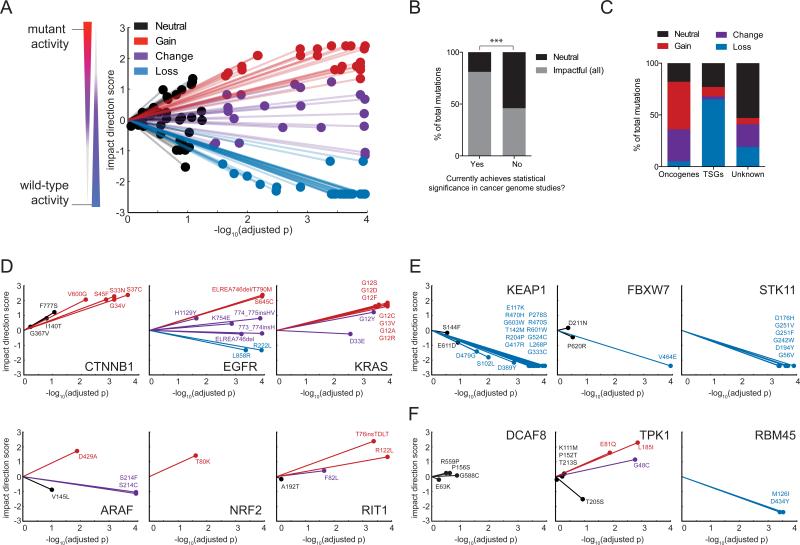
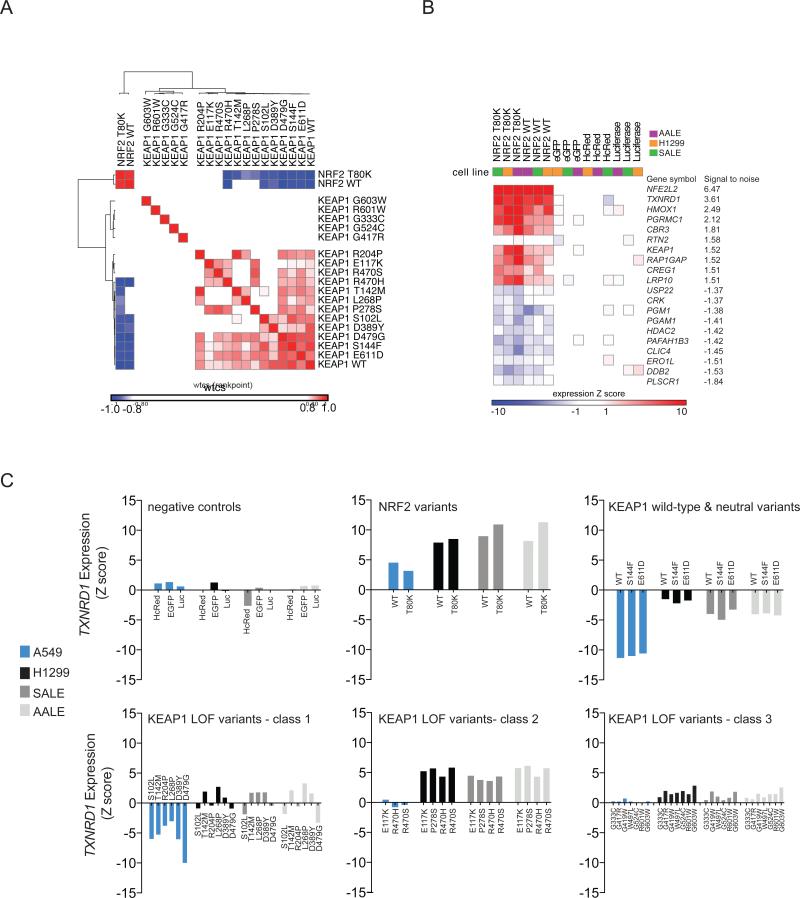
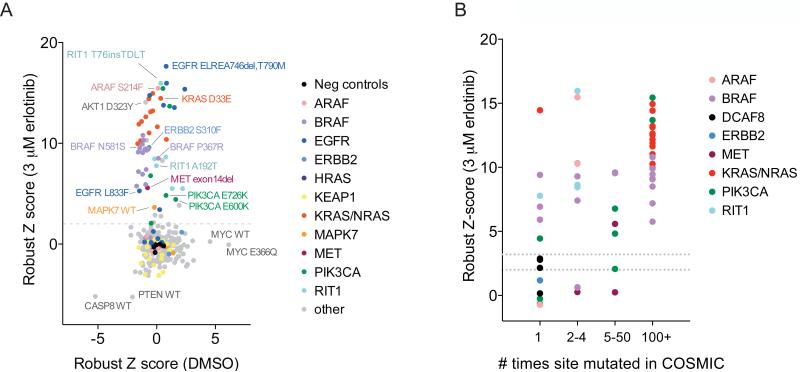
Comment in
-
Functionally Assessing Candidate Drivers Advances Precision Cancer Medicine.Cancer Cell. 2016 Aug 8;30(2):187-189. doi: 10.1016/j.ccell.2016.07.011. Cancer Cell. 2016. PMID: 27505666
-
Lung cancer: Functional insights into cancer mutations-eVIP approach.Nat Rev Clin Oncol. 2016 Aug 19;13(9):527. doi: 10.1038/nrclinonc.2016.131. Nat Rev Clin Oncol. 2016. PMID: 27538976 No abstract available.
References
-
- Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004;428:431–437. - PubMed
-
- Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, Sjostrom SK, Garraway LA, Weremowicz S, Richardson AL, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–1079. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous