

Emerging concepts of epigenetic dysregulation in hematological malignancies
- PMID: 27478938
- PMCID: PMC5134743
- DOI: 10.1038/ni.3517
Emerging concepts of epigenetic dysregulation in hematological malignancies
Abstract
The past decade brought a revolution in understanding of the structure, topology and disease-inducing lesions of RNA and DNA, fueled by unprecedented progress in next-generation sequencing. This technological revolution has also affected understanding of the epigenome and has provided unique opportunities for the analysis of DNA and histone modifications, as well as the first map of the non-protein-coding genome and three-dimensional (3D) chromosomal interactions. Overall, these advances have facilitated studies that combine genetic, transcriptomics and epigenomics data to address a wide range of issues ranging from understanding the role of the epigenome in development to targeting the transcription of noncoding genes in human cancer. Here we describe recent insights into epigenetic dysregulation characteristic of the malignant differentiation of blood stem cells based on studies of alterations that affect epigenetic complexes, enhancers, chromatin, long noncoding RNAs (lncRNAs), RNA splicing, nuclear topology and the 3D conformation of chromatin.
Figures
References
-
- Heintzman ND, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007;39:311–318. - PubMed
-
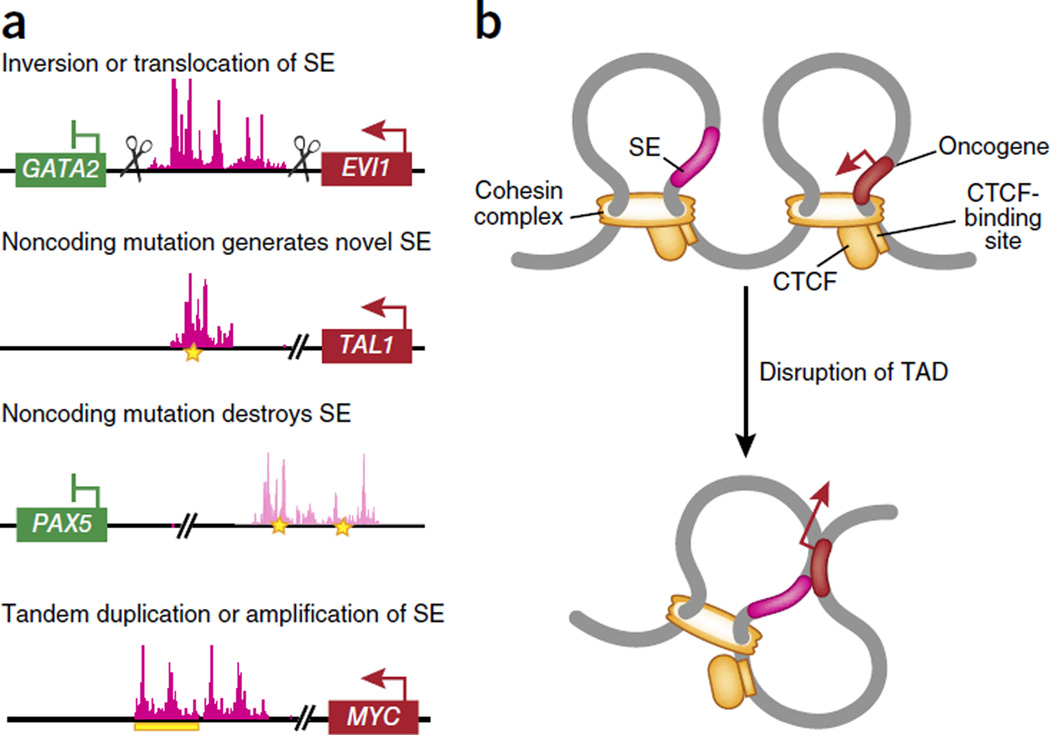
- Gröschel S, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157:369–381. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA173636/CA/NCI NIH HHS/United States
- R01 CA133379/CA/NCI NIH HHS/United States
- K08 CA160647/CA/NCI NIH HHS/United States
- R01 CA149655/CA/NCI NIH HHS/United States
- R01 CA105129/CA/NCI NIH HHS/United States
- R01 HL128239/HL/NHLBI NIH HHS/United States
- P30 CA060553/CA/NCI NIH HHS/United States
- R01 CA202025/CA/NCI NIH HHS/United States
- R01 CA169784/CA/NCI NIH HHS/United States
- R00 CA188293/CA/NCI NIH HHS/United States
- R01 CA190509/CA/NCI NIH HHS/United States
- R01 CA194923/CA/NCI NIH HHS/United States
- R01 CA202027/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
