

Tempo and mode of genome evolution in a 50,000-generation experiment
- PMID: 27479321
- PMCID: PMC4988878
- DOI: 10.1038/nature18959
Tempo and mode of genome evolution in a 50,000-generation experiment
Abstract
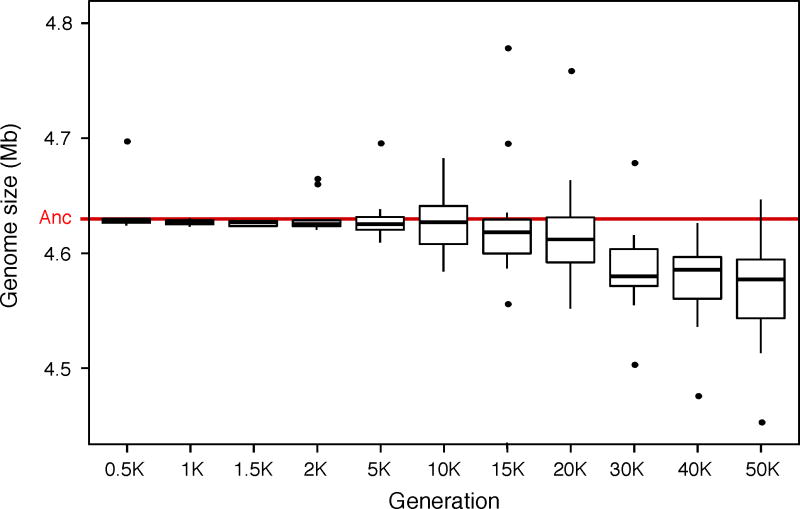
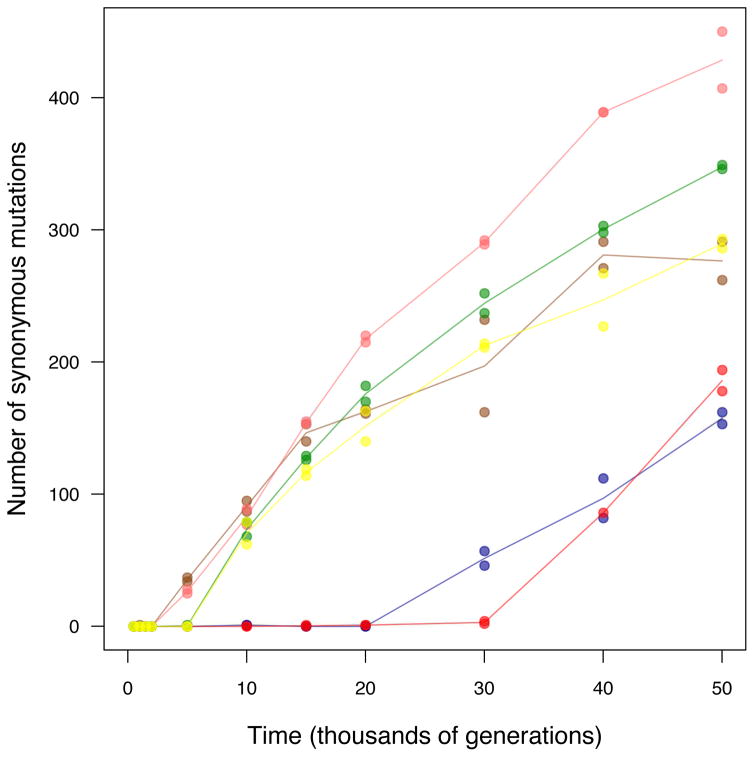
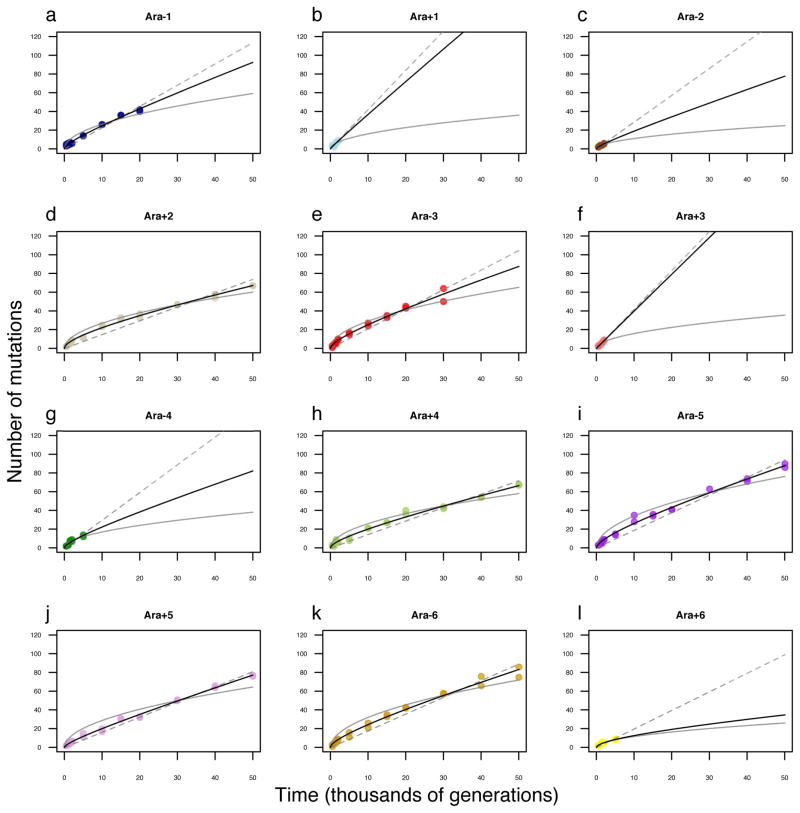
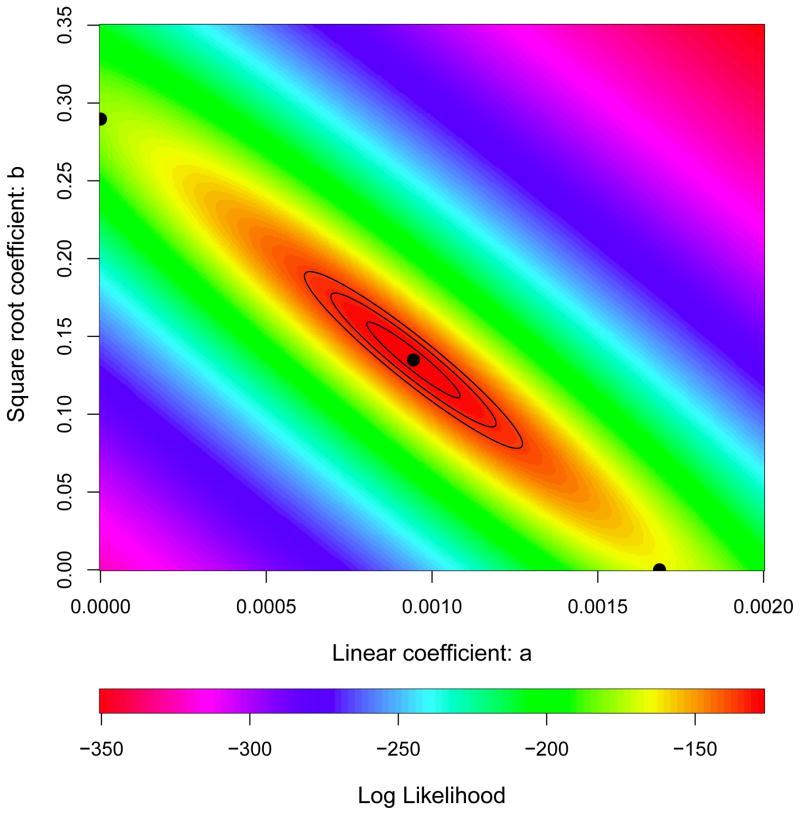
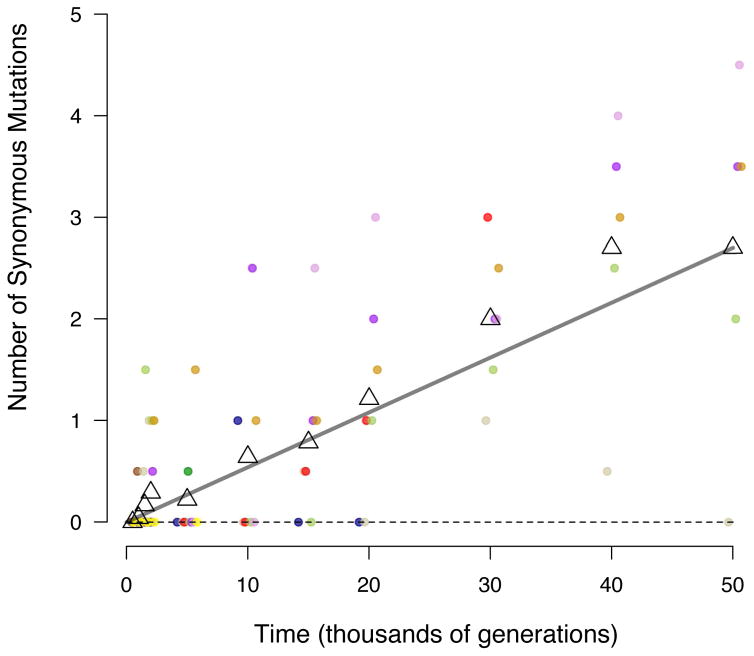
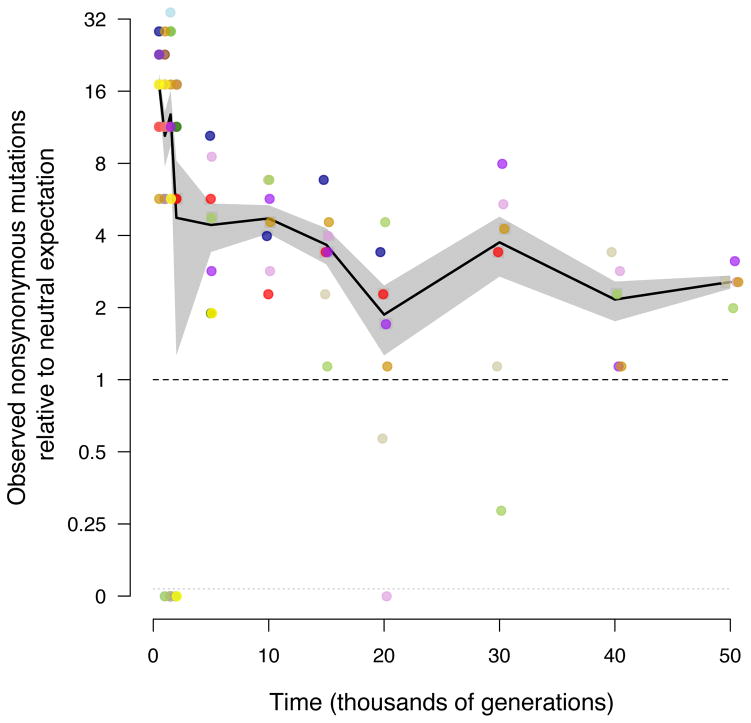
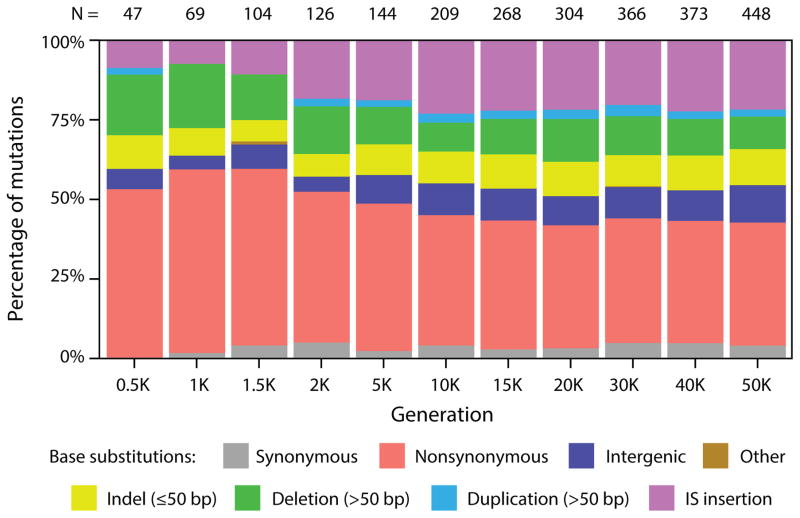
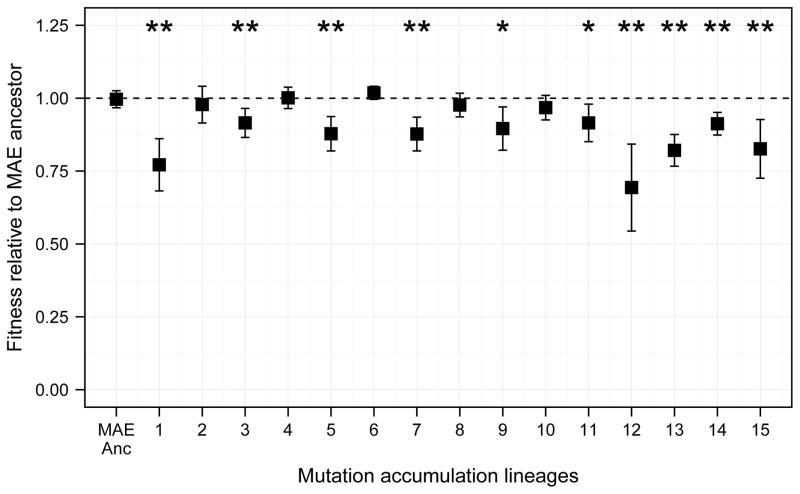
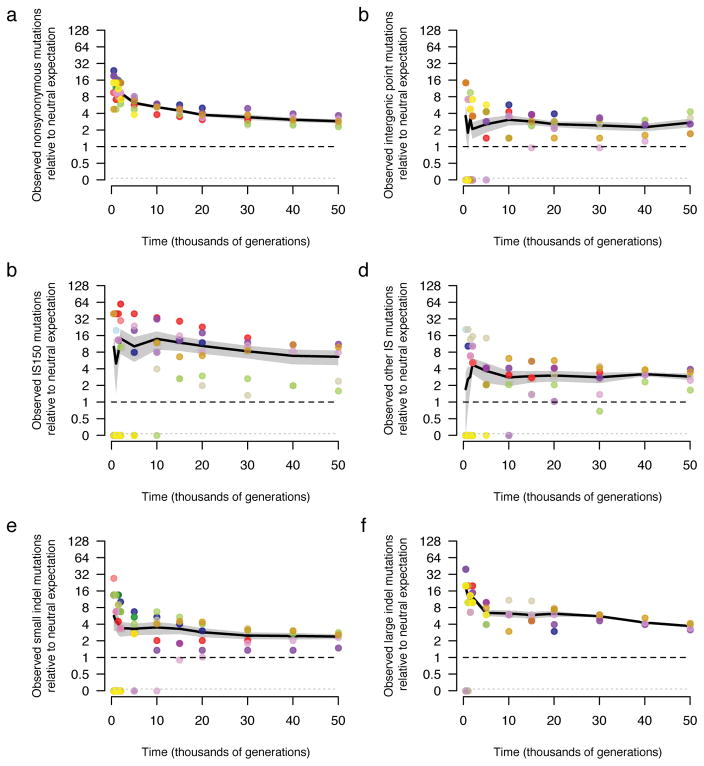
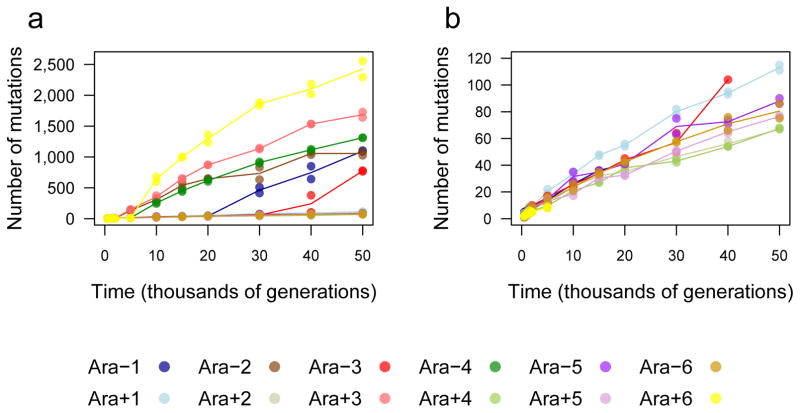
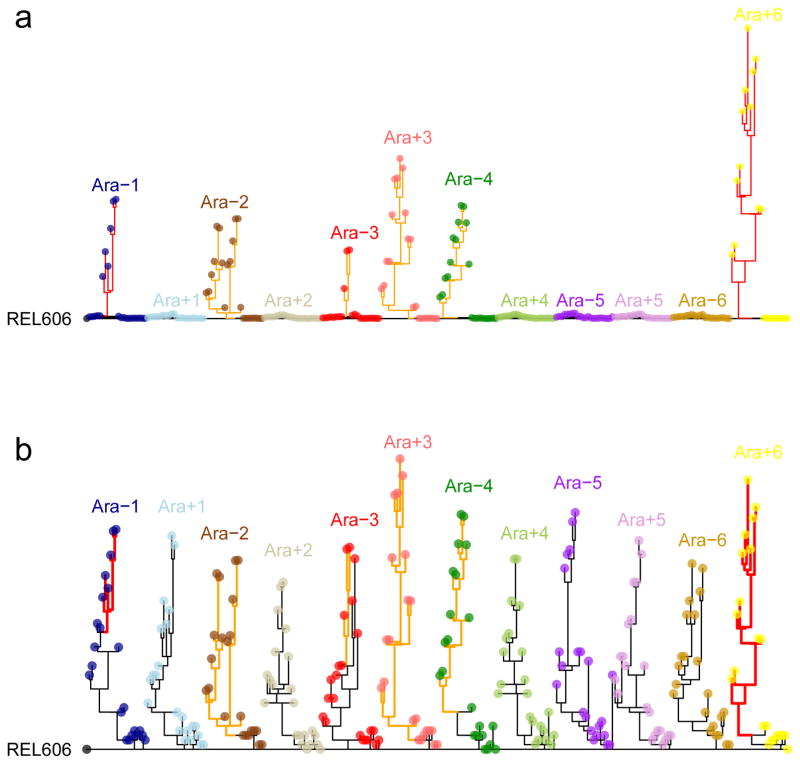
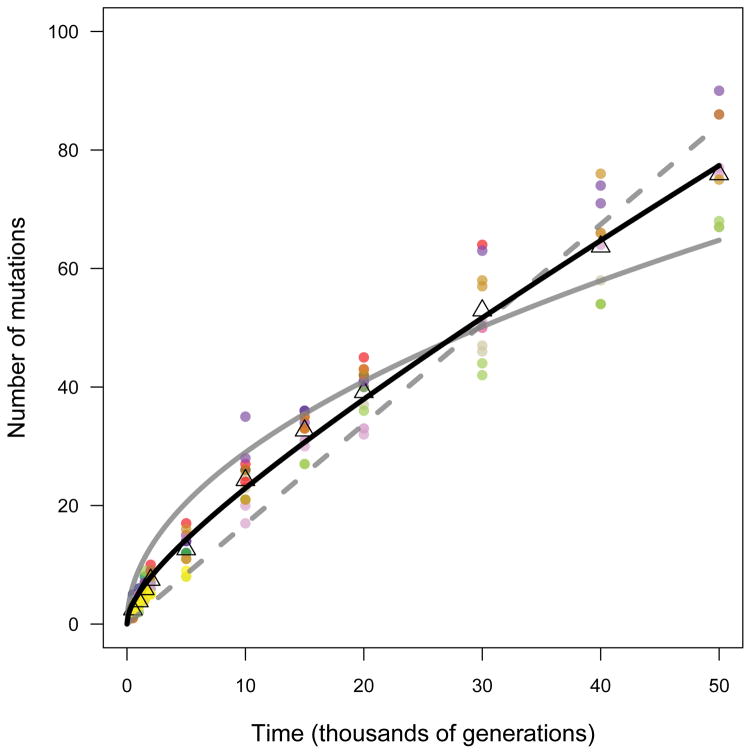
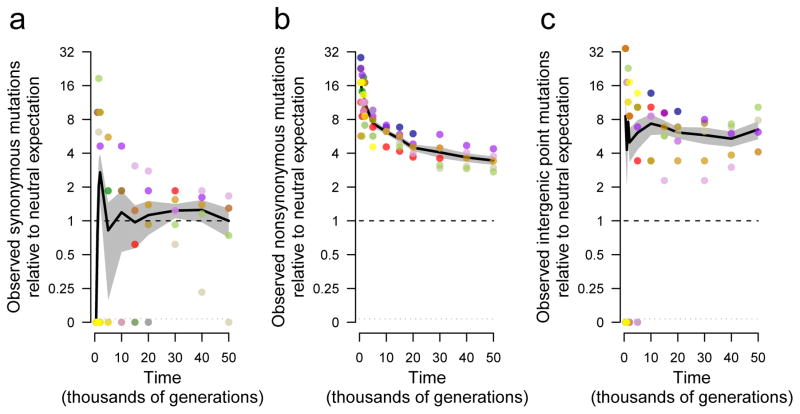
Adaptation by natural selection depends on the rates, effects and interactions of many mutations, making it difficult to determine what proportion of mutations in an evolving lineage are beneficial. Here we analysed 264 complete genomes from 12 Escherichia coli populations to characterize their dynamics over 50,000 generations. The populations that retained the ancestral mutation rate support a model in which most fixed mutations are beneficial, the fraction of beneficial mutations declines as fitness rises, and neutral mutations accumulate at a constant rate. We also compared these populations to mutation-accumulation lines evolved under a bottlenecking regime that minimizes selection. Nonsynonymous mutations, intergenic mutations, insertions and deletions are overrepresented in the long-term populations, further supporting the inference that most mutations that reached high frequency were favoured by selection. These results illuminate the shifting balance of forces that govern genome evolution in populations adapting to a new environment.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. - PubMed
