

Profibrotic up-regulation of glucose transporter 1 by TGF-β involves activation of MEK and mammalian target of rapamycin complex 2 pathways
- PMID: 27480571
- PMCID: PMC5067255
- DOI: 10.1096/fj.201600428R
Profibrotic up-regulation of glucose transporter 1 by TGF-β involves activation of MEK and mammalian target of rapamycin complex 2 pathways
Abstract
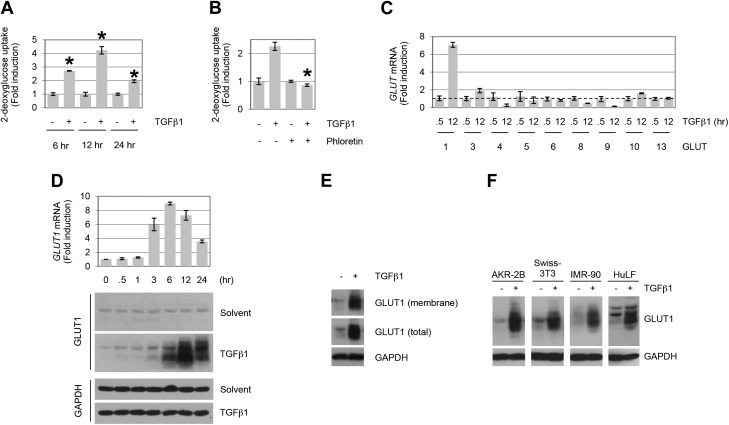
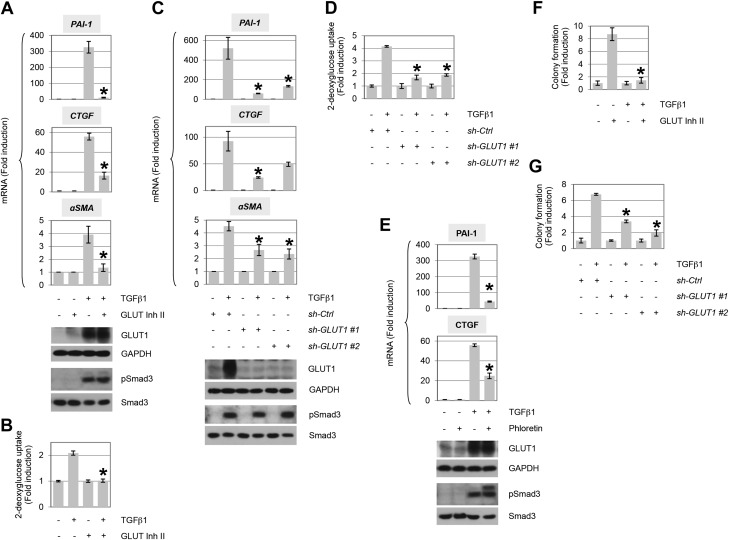
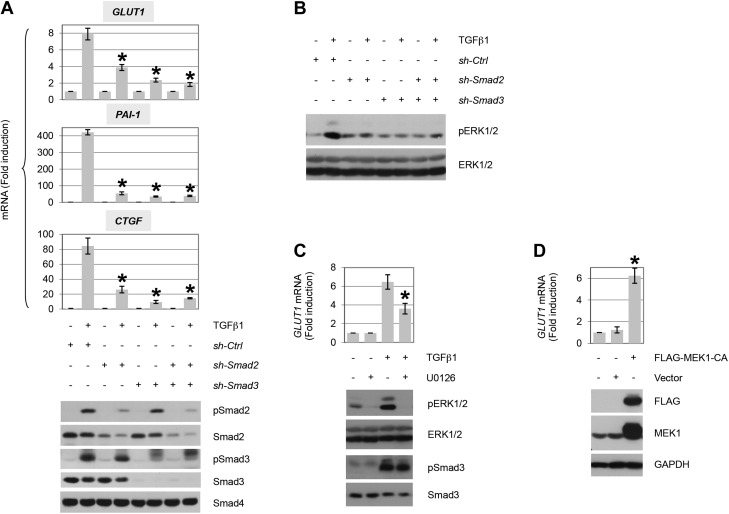
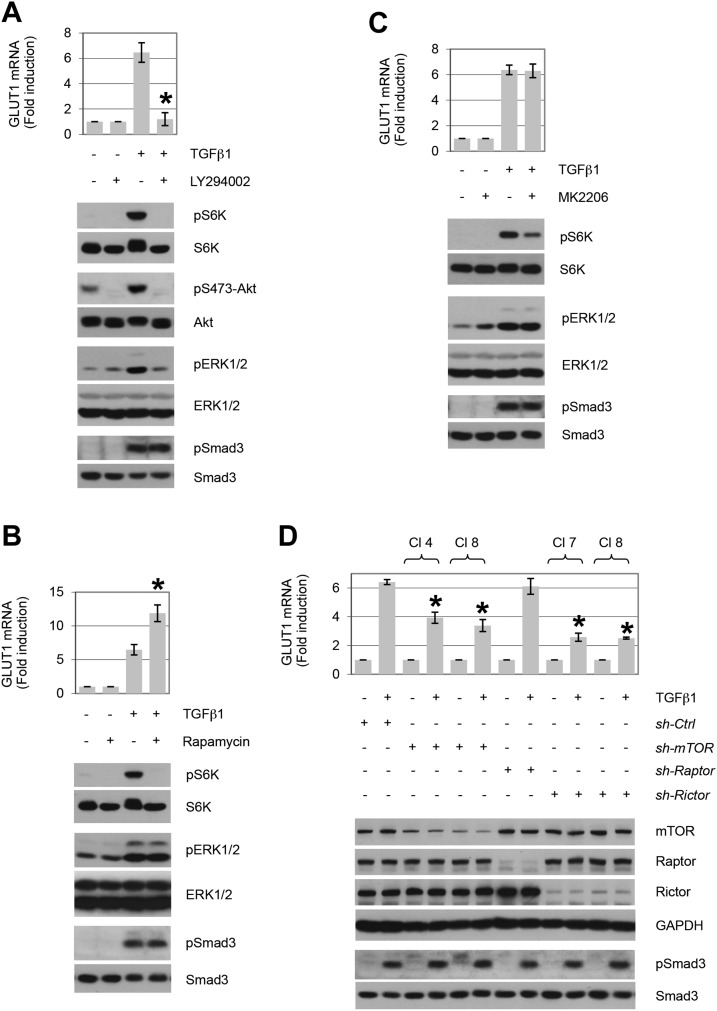
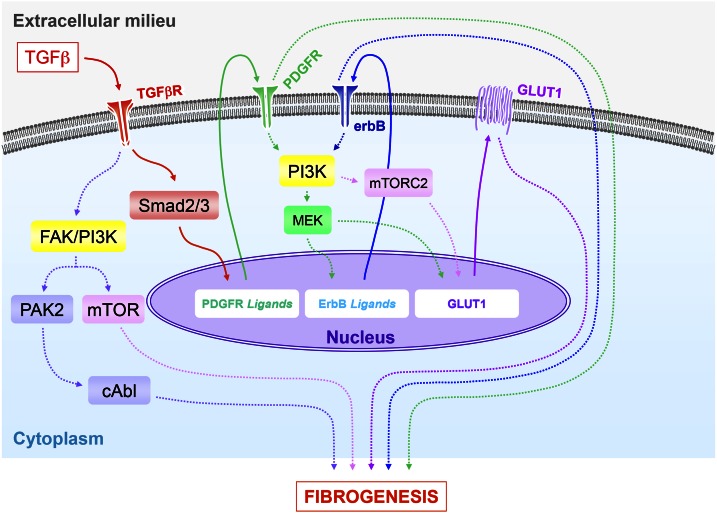
TGF-β plays a central role in the pathogenesis of fibroproliferative disorders. Defining the exact underlying molecular basis is therefore critical for the development of viable therapeutic strategies. Here, we show that expression of the facilitative glucose transporter 1 (GLUT1) is induced by TGF-β in fibroblast lines and primary cells and is required for the profibrotic effects of TGF-β. In addition, enhanced GLUT1 expression is observed in fibrotic areas of lungs of both patients with idiopathic pulmonary fibrosis and mice that are subjected to a fibrosis-inducing bleomycin treatment. By using pharmacologic and genetic approaches, we demonstrate that up-regulation of GLUT1 occurs via the canonical Smad2/3 pathway and requires autocrine activation of the receptor tyrosine kinases, platelet-derived and epidermal growth factor receptors. Engagement of the common downstream effector PI3K subsequently triggers activation of the MEK and mammalian target of rapamycin complex 2, which cooperate in regulating GLUT1 expression. Of note, inhibition of GLUT1 activity and/or expression is shown to impair TGF-β-driven fibrogenic processes, including cell proliferation and production of profibrotic mediators. These findings provide new perspectives on the interrelation of metabolism and profibrotic TGF-β signaling and present opportunities for potential therapeutic intervention.-Andrianifahanana, M., Hernandez, D. M., Yin, X., Kang, J.-H., Jung, M.-Y., Wang, Y., Yi, E. S., Roden, A. C., Limper, A. H., Leof, E. B. Profibrotic up-regulation of glucose transporter 1 by TGF-β involves activation of MEK and mammalian target of rapamycin complex 2 pathways.
Keywords: fibrosis; metabolism; signaling.
© FASEB.
Figures


References
-
- Pinzani M., Vizzutti F. (2008) Fibrosis and cirrhosis reversibility: clinical features and implications. Clin. Liver Dis. 12, 901–913 - PubMed
-
- Sivakumar P., Ntolios P., Jenkins G., Laurent G. (2012) Into the matrix: targeting fibroblasts in pulmonary fibrosis. Curr. Opin. Pulm. Med. 18, 462–469 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
