

Beneficial Role of Erythrocyte Adenosine A2B Receptor-Mediated AMP-Activated Protein Kinase Activation in High-Altitude Hypoxia
- PMID: 27482003
- PMCID: PMC6168195
- DOI: 10.1161/CIRCULATIONAHA.116.021311
Beneficial Role of Erythrocyte Adenosine A2B Receptor-Mediated AMP-Activated Protein Kinase Activation in High-Altitude Hypoxia
Abstract
Background: High altitude is a challenging condition caused by insufficient oxygen supply. Inability to adjust to hypoxia may lead to pulmonary edema, stroke, cardiovascular dysfunction, and even death. Thus, understanding the molecular basis of adaptation to high altitude may reveal novel therapeutics to counteract the detrimental consequences of hypoxia.
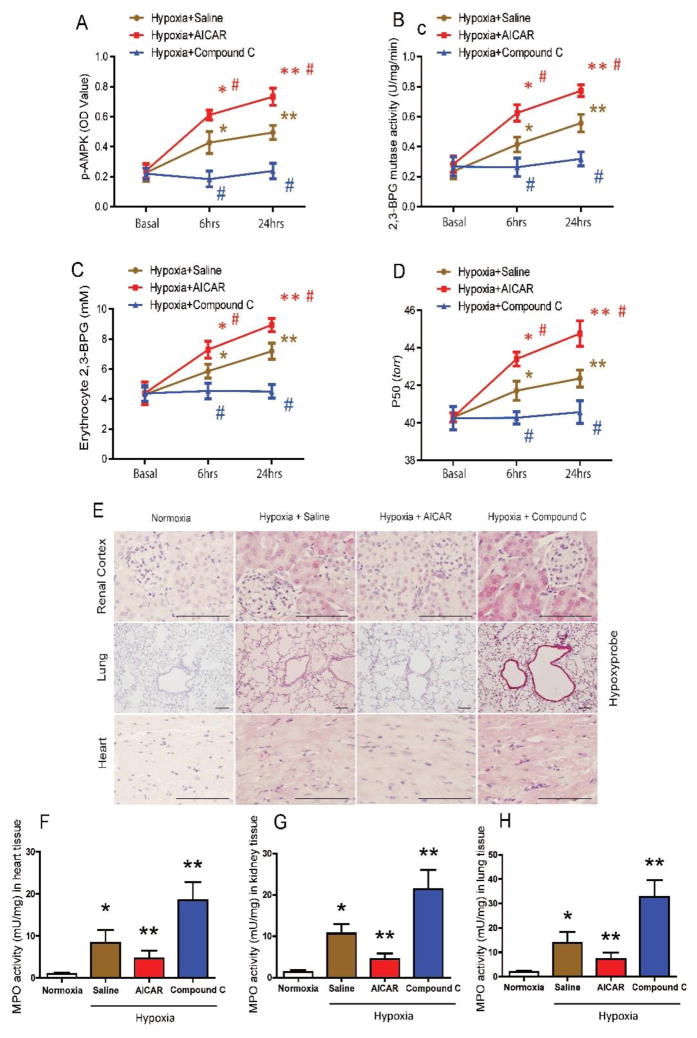
Methods: Using high-throughput, unbiased metabolomic profiling, we report that the metabolic pathway responsible for production of erythrocyte 2,3-bisphosphoglycerate (2,3-BPG), a negative allosteric regulator of hemoglobin-O2 binding affinity, was significantly induced in 21 healthy humans within 2 hours of arrival at 5260 m and further increased after 16 days at 5260 m.
Results: This finding led us to discover that plasma adenosine concentrations and soluble CD73 activity rapidly increased at high altitude and were associated with elevated erythrocyte 2,3-BPG levels and O2 releasing capacity. Mouse genetic studies demonstrated that elevated CD73 contributed to hypoxia-induced adenosine accumulation and that elevated adenosine-mediated erythrocyte A2B adenosine receptor activation was beneficial by inducing 2,3-BPG production and triggering O2 release to prevent multiple tissue hypoxia, inflammation, and pulmonary vascular leakage. Mechanistically, we demonstrated that erythrocyte AMP-activated protein kinase was activated in humans at high altitude and that AMP-activated protein kinase is a key protein functioning downstream of the A2B adenosine receptor, phosphorylating and activating BPG mutase and thus inducing 2,3-BPG production and O2 release from erythrocytes. Significantly, preclinical studies demonstrated that activation of AMP-activated protein kinase enhanced BPG mutase activation, 2,3-BPG production, and O2 release capacity in CD73-deficient mice, in erythrocyte-specific A2B adenosine receptor knockouts, and in wild-type mice and in turn reduced tissue hypoxia and inflammation.
Conclusions: Together, human and mouse studies reveal novel mechanisms of hypoxia adaptation and potential therapeutic approaches for counteracting hypoxia-induced tissue damage.
Keywords: AMP-activated protein kinases; adenosine; altitude; hypoxia, brain; oxygen; signal transduction.
© 2016 American Heart Association, Inc.
Figures





References
-
- Anderson JD, Honigman B. The effect of altitude-induced hypoxia on heart disease: Do acute, intermittent, and chronic exposures provide cardioprotection? High Alt Med Biol. 2011;12:45–55. - PubMed
-
- Weissmann N, Sommer N, Schermuly RT, Ghofrani HA, Seeger W, Grimminger F. Oxygen sensors in hypoxic pulmonary vasoconstriction. Cardiovasc Res. 2006;71:620–629. - PubMed
-
- Tuder RM, Yun JH, Bhunia A, Fijalkowska I. Hypoxia and chronic lung disease. J Mol Med (Berl) 2007;85:1317–1324. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
