

Distinct cellular properties of oncogenic KIT receptor tyrosine kinase mutants enable alternative courses of cancer cell inhibition
- PMID: 27482095
- PMCID: PMC4995958
- DOI: 10.1073/pnas.1610179113
Distinct cellular properties of oncogenic KIT receptor tyrosine kinase mutants enable alternative courses of cancer cell inhibition
Abstract
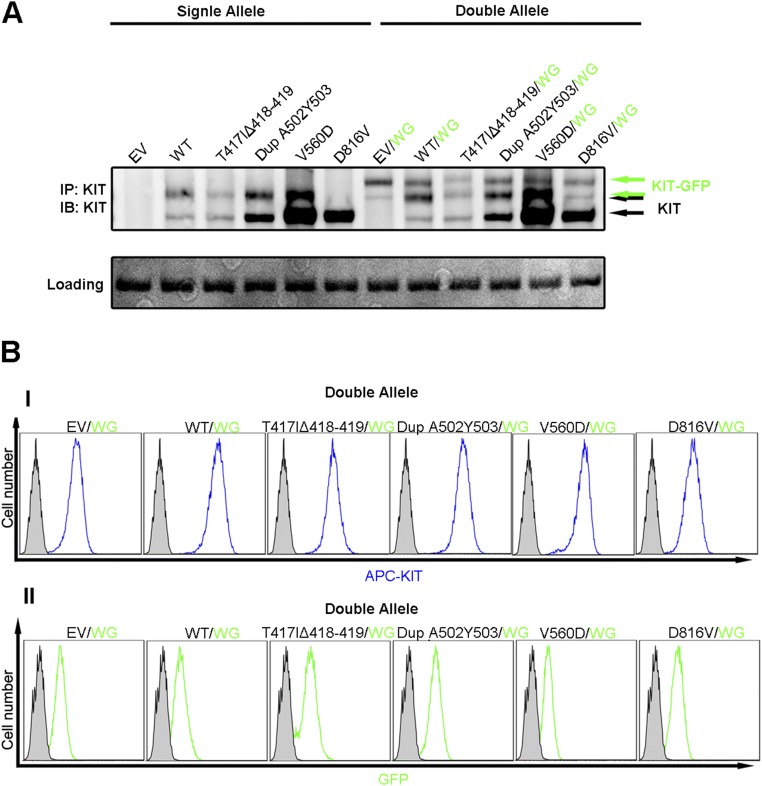
Large genomic sequencing analysis as part of precision medicine efforts revealed numerous activating mutations in receptor tyrosine kinases, including KIT. Unfortunately, a single approach is not effective for inhibiting cancer cells or treating cancers driven by all known oncogenic KIT mutants. Here, we show that each of the six major KIT oncogenic mutants exhibits different enzymatic, cellular, and dynamic properties and responds distinctly to different KIT inhibitors. One class of KIT mutants responded well to anti-KIT antibody treatment alone or in combination with a low dose of tyrosine kinase inhibitors (TKIs). A second class of KIT mutants, including a mutant resistant to imatinib treatment, responded well to a combination of TKI with anti-KIT antibodies or to anti-KIT toxin conjugates, respectively. We conclude that the preferred choice of precision medicine treatments for cancers driven by activated KIT and other RTKs may rely on clear understanding of the dynamic properties of oncogenic mutants.
Keywords: KIT; monoclonal antibody; oncogenic mutant; receptor tyrosine kinase; targeted therapy.
Conflict of interest statement
E.M.M.-B. and Y.H. are employees of Kolltan Pharmaceuticals.
Figures













References
-
- Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11(10):685–696. - PubMed
-
- Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153(1):17–37. - PubMed
-
- Ashman LK, Griffith R. Therapeutic targeting of c-KIT in cancer. Expert Opin Investig Drugs. 2013;22(1):103–115. - PubMed
-
- Ashman LK. The biology of stem cell factor and its receptor C-kit. Int J Biochem Cell Biol. 1999;31(10):1037–1051. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
