

Expanding the clinical spectrum of COL1A1 mutations in different forms of glaucoma
- PMID: 27484908
- PMCID: PMC4970237
- DOI: 10.1186/s13023-016-0495-y
Expanding the clinical spectrum of COL1A1 mutations in different forms of glaucoma
Abstract
Background: Primary congenital glaucoma (PCG) and early onset glaucomas are one of the major causes of children and young adult blindness worldwide. Both autosomal recessive and dominant inheritance have been described with involvement of several genes including CYP1B1, FOXC1, PITX2, MYOC and PAX6. However, mutations in these genes explain only a small fraction of cases suggesting the presence of further candidate genes.
Methods: To elucidate further genetic causes of these conditions whole exome sequencing (WES) was performed in an Italian patient, diagnosed with PCG and retinal detachment, and his unaffected parents. Sanger sequencing of the complete coding region of COL1A1 was performed in a total of 26 further patients diagnosed with PCG or early onset glaucoma. Exclusion of pathogenic variations in known glaucoma genes as CYP1B1, MYOC, FOXC1, PITX2 and PAX6 was additionally done per Sanger sequencing and Multiple Ligation-dependent Probe Amplification (MLPA) analysis.
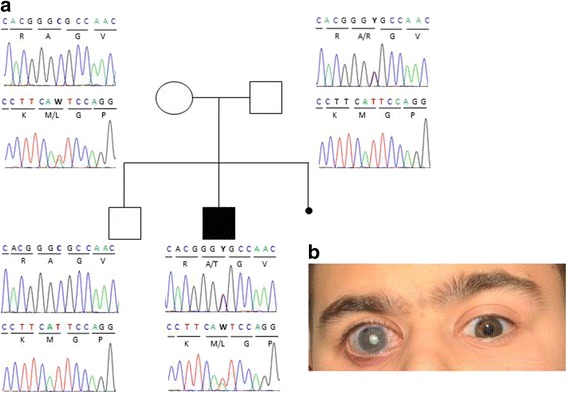
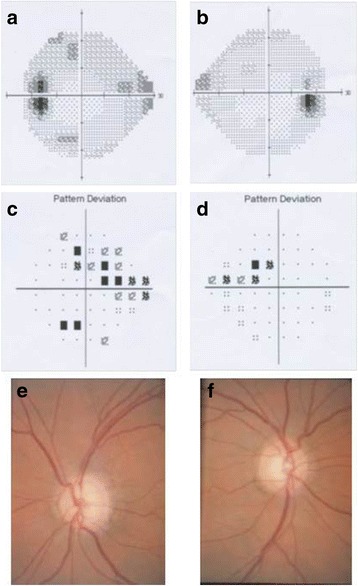
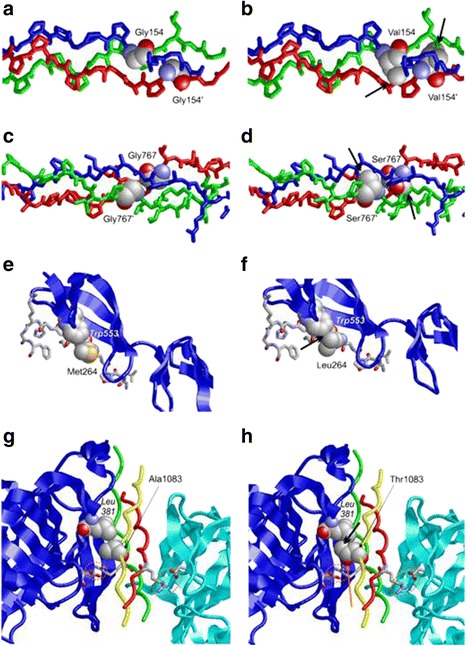
Results: In the patient diagnosed with PCG and retinal detachment, analysis of WES data identified compound heterozygous variants in COL1A1 (p.Met264Leu; p.Ala1083Thr). Targeted COL1A1 screening of 26 additional patients detected three further heterozygous variants (p.Arg253*, p.Gly767Ser and p.Gly154Val) in three distinct subjects: two of them diagnosed with early onset glaucoma and mild form of osteogenesis imperfecta (OI), one patient with a diagnosis of PCG at age 4 years. All five variants affected evolutionary, highly conserved amino acids indicating important functional restrictions. Molecular modeling predicted that the heterozygous variants are dominant in effect and affect protein stability and thus the amount of available protein, while the compound heterozygous variants act as recessive alleles and impair binding affinity to two main COL1A1 binding proteins: Hsp47 and fibronectin.
Conclusions: Dominant inherited mutations in COL1A1 are known causes of connective tissues disorders such as OI. These disorders are also associated with different ocular abnormalities, although recognition of the common pathology for both features is seldom being recognized. Our results expand the role of COL1A1 mutations in different forms of early-onset glaucoma with and without signs of OI. Thus, we suggest including COL1A1 mutation screening in the genetic work-up of glaucoma cases and detailed ophthalmic examinations with fundus analysis in patients with OI.
Keywords: COL1A1; Congenital glaucoma; Early onset glaucoma; Osteogenesis imperfecta; Whole exome sequencing.
Figures

Similar articles
-
Molecular characterization of Axenfeld-Rieger spectrum and other anterior segment dysgeneses in a sample of Mexican patients.Ophthalmic Genet. 2018 Dec;39(6):728-734. doi: 10.1080/13816810.2018.1547911. Epub 2018 Nov 20. Ophthalmic Genet. 2018. PMID: 30457409
-
Mutations of CYP1B1 and FOXC1 genes for childhood glaucoma in Japanese individuals.Jpn J Ophthalmol. 2024 Nov;68(6):688-701. doi: 10.1007/s10384-024-01103-0. Epub 2024 Aug 19. Jpn J Ophthalmol. 2024. PMID: 39158757 Free PMC article.
-
Digenic inheritance of early-onset glaucoma: CYP1B1, a potential modifier gene.Am J Hum Genet. 2002 Feb;70(2):448-60. doi: 10.1086/338709. Epub 2002 Jan 3. Am J Hum Genet. 2002. PMID: 11774072 Free PMC article.
-
Primary congenital and developmental glaucomas.Hum Mol Genet. 2017 Aug 1;26(R1):R28-R36. doi: 10.1093/hmg/ddx205. Hum Mol Genet. 2017. PMID: 28549150 Free PMC article. Review.
-
Rare splicing mutation in COL1A1 gene identified by whole exomes sequencing in a patient with osteogenesis imperfecta type I followed by prenatal diagnosis: A case report and review of the literature.Gene. 2020 May 30;741:144565. doi: 10.1016/j.gene.2020.144565. Epub 2020 Mar 10. Gene. 2020. PMID: 32165296
Cited by
-
An Assessment of GUCA1C Variants in Primary Congenital Glaucoma.Genes (Basel). 2021 Mar 2;12(3):359. doi: 10.3390/genes12030359. Genes (Basel). 2021. PMID: 33801495 Free PMC article.
-
Abnormal corneal properties in osteogenesis imperfecta and glaucoma: a case series.BMJ Open Ophthalmol. 2021 Apr 15;6(1):e000684. doi: 10.1136/bmjophth-2020-000684. eCollection 2021. BMJ Open Ophthalmol. 2021. PMID: 33928192 Free PMC article.
-
Keratoconus tomographic indices in osteogenesis imperfecta.Graefes Arch Clin Exp Ophthalmol. 2023 Sep;261(9):2585-2592. doi: 10.1007/s00417-023-06059-4. Epub 2023 Apr 19. Graefes Arch Clin Exp Ophthalmol. 2023. PMID: 37074408 Free PMC article.
-
Generation of a Matrix Gla (Mgp) floxed mouse, followed by conditional knockout, uncovers a new Mgp function in the eye.Sci Rep. 2020 Oct 29;10(1):18583. doi: 10.1038/s41598-020-75031-7. Sci Rep. 2020. PMID: 33122788 Free PMC article.
-
PITX2 deficiency and associated human disease: insights from the zebrafish model.Hum Mol Genet. 2018 May 15;27(10):1675-1695. doi: 10.1093/hmg/ddy074. Hum Mol Genet. 2018. PMID: 29506241 Free PMC article.
References
-
- Stoilov I, Akarsu AN, Sarfarazi M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (Buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum Mol Genet. 1997;6(4):641–7. doi: 10.1093/hmg/6.4.641. - DOI - PubMed
-
- Pasutto F, Chavarria-Soley G, Mardin CY, Michels-Rautenstrauss K, Ingelman-Sundberg M, Fernandez-Martinez L, Weber BH, Rautenstrauss B, Reis A. Heterozygous loss-of-function variants in CYP1B1 predispose to primary open-angle glaucoma. Invest Ophthalmol Vis Sci. 2010;51(1):249–54. doi: 10.1167/iovs.09-3880. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
