

Rasip1-Mediated Rho GTPase Signaling Regulates Blood Vessel Tubulogenesis via Nonmuscle Myosin II
- PMID: 27486147
- PMCID: PMC5026621
- DOI: 10.1161/CIRCRESAHA.116.309094
Rasip1-Mediated Rho GTPase Signaling Regulates Blood Vessel Tubulogenesis via Nonmuscle Myosin II
Abstract
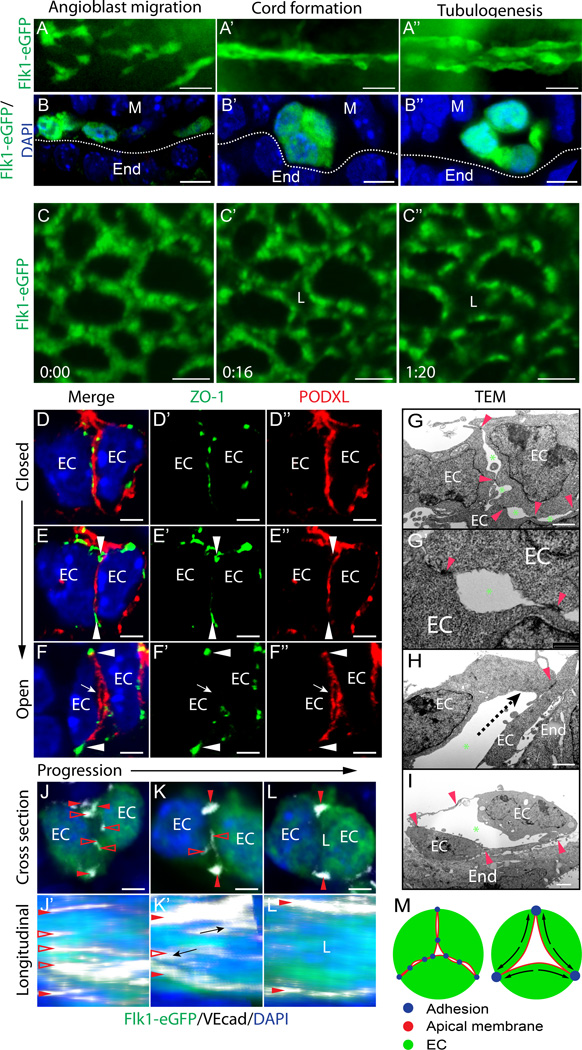
Rationale: Vascular tubulogenesis is essential to cardiovascular development. Within initial vascular cords of endothelial cells, apical membranes are established and become cleared of cell-cell junctions, thereby allowing continuous central lumens to open. Rasip1 (Ras-interacting protein 1) is required for apical junction clearance, as well as for regulation of Rho GTPase (enzyme that hydrolyzes GTP) activity. However, it remains unknown how activities of different Rho GTPases are coordinated by Rasip1 to direct tubulogenesis.
Objective: The aim of this study is to determine the mechanisms downstream of Rasip1 that drive vascular tubulogenesis.
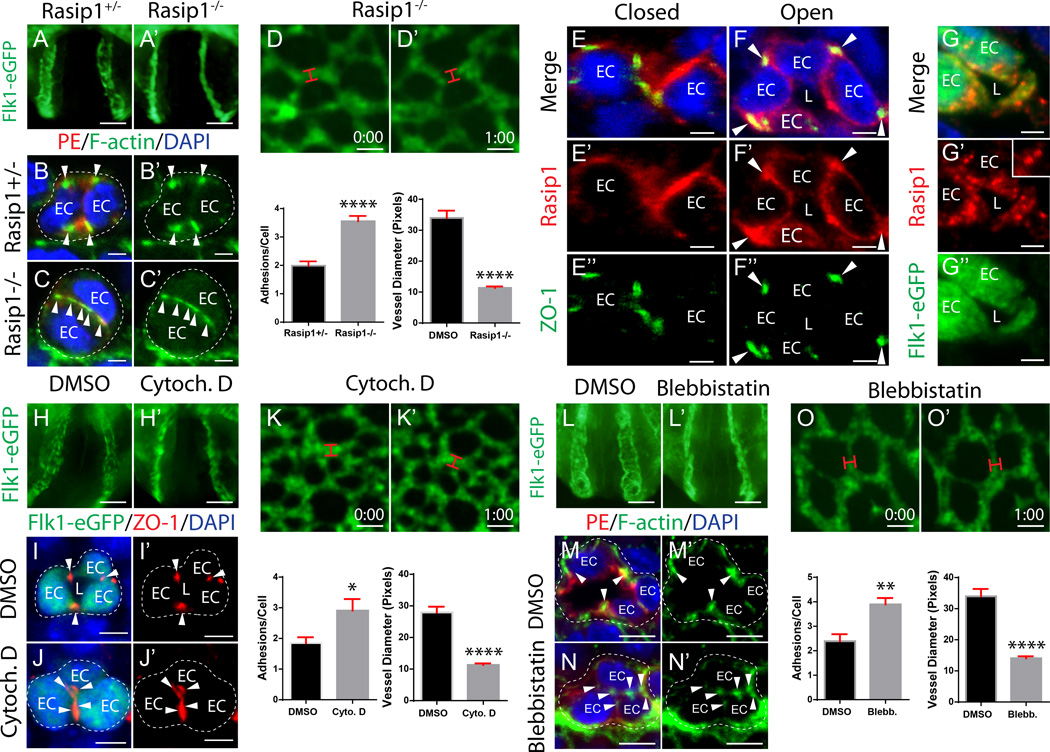
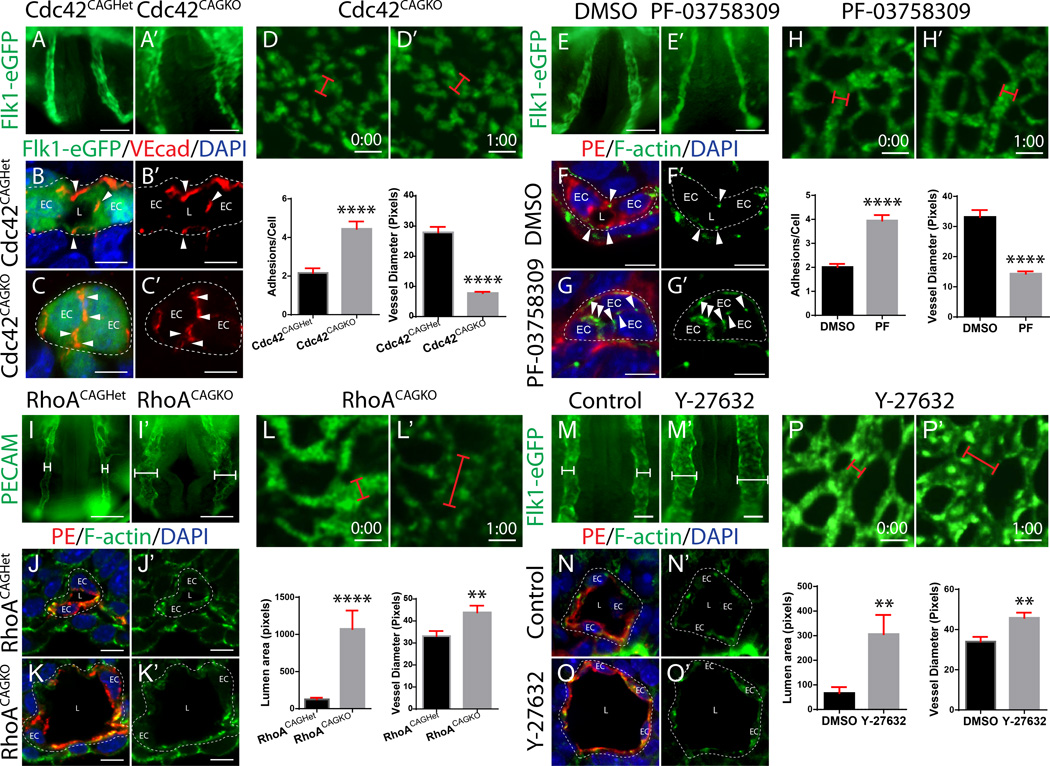
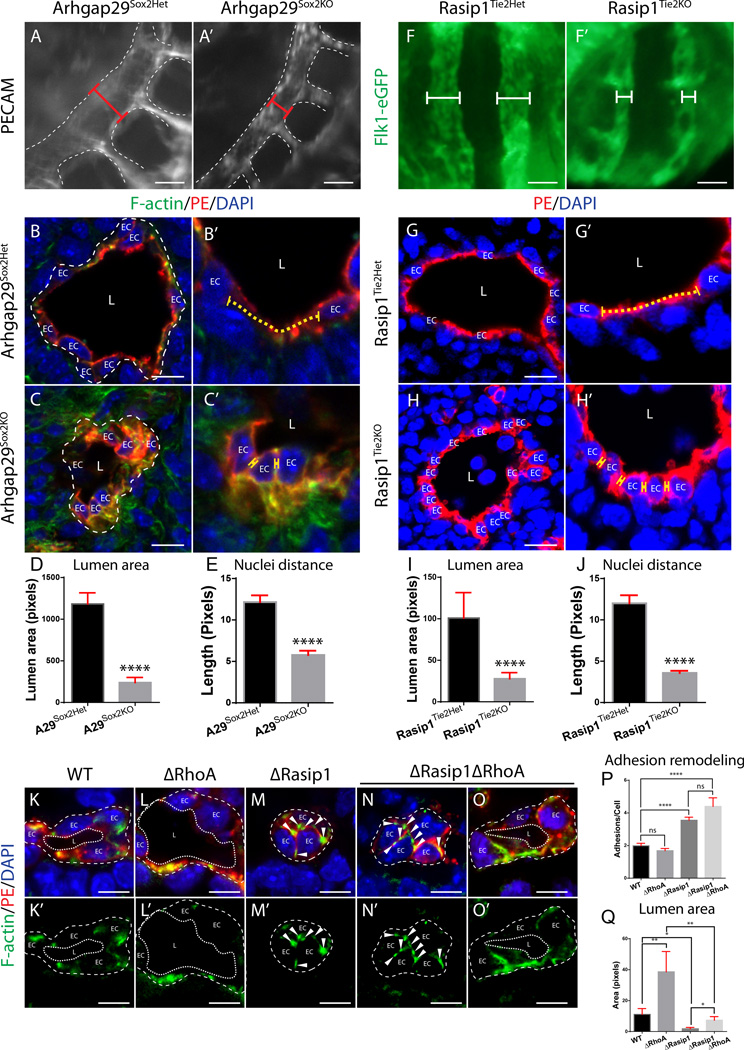
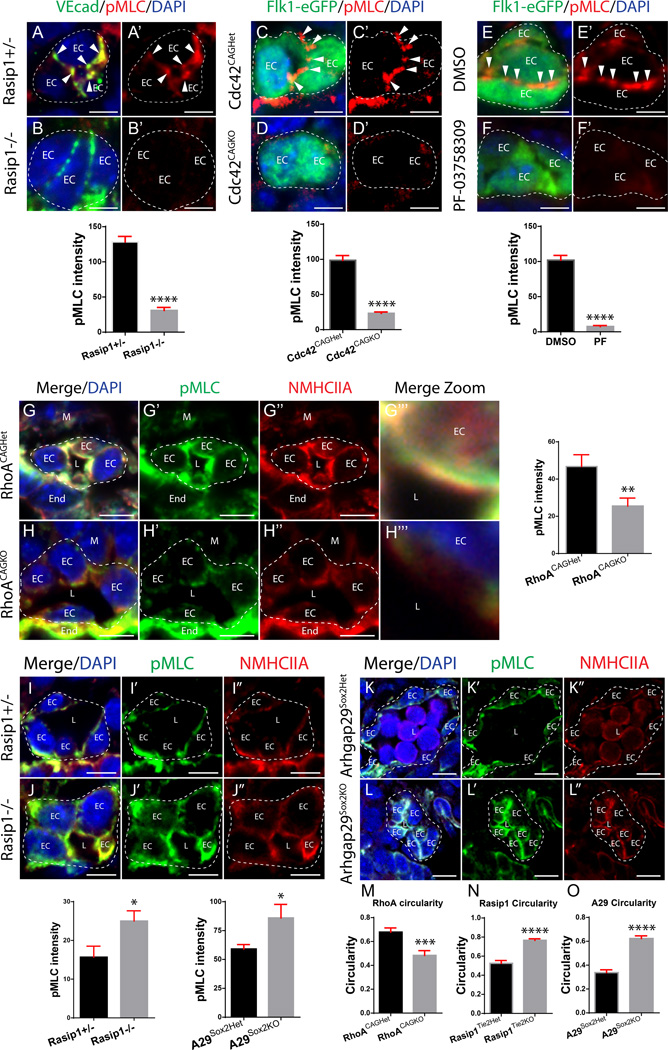
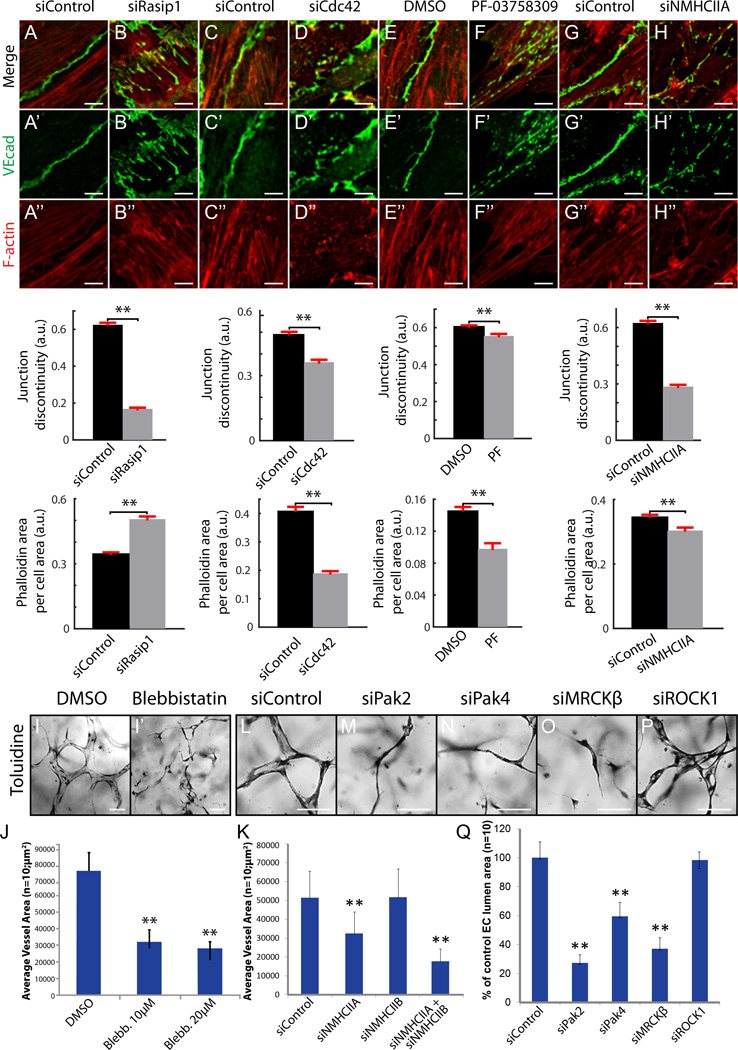
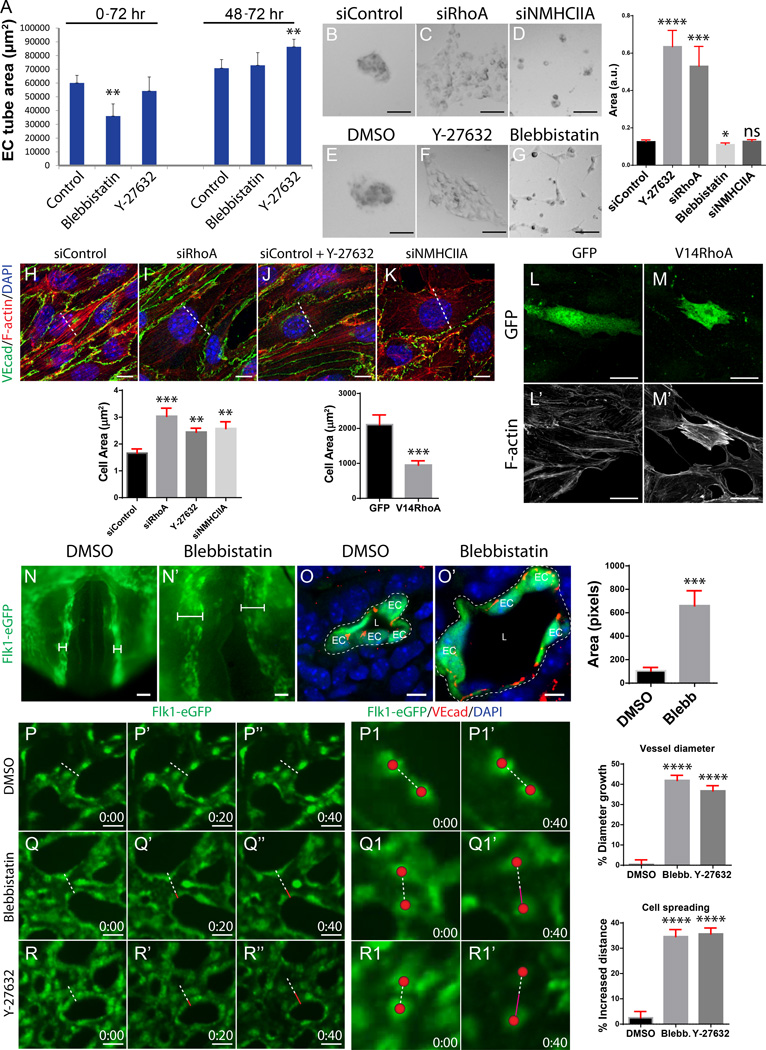
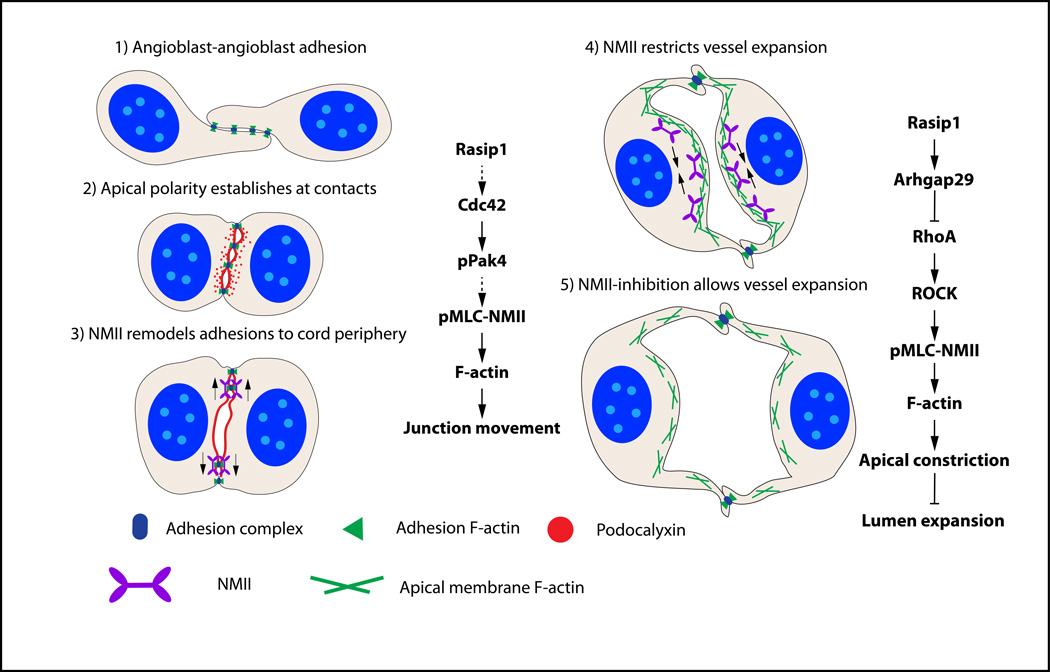
Methods and results: Using conditional mouse mutant models and pharmacological approaches, we dissect GTPase pathways downstream of Rasip1. We show that clearance of endothelial cell apical junctions during vascular tubulogenesis depends on Rasip1, as well as the GTPase Cdc42 (cell division control protein 42 homolog) and the kinase Pak4 (serine/threonine-protein kinase 4). Genetic deletion of Rasip1 or Cdc42, or inhibition of Pak4, all blocks endothelial cell tubulogenesis. By contrast, inactivation of RhoA (Ras homologue gene family member A) signaling leads to vessel overexpansion, implicating actomyosin contractility in control of lumen diameter. Interestingly, blocking activity of NMII (nonmuscle myosin II) either before, or after, lumen morphogenesis results in dramatically different tubulogenesis phenotypes, suggesting time-dependent roles.
Conclusions: Rasip1 controls different pools of GTPases, which in turn regulate different pools of NMII to coordinate junction clearance (remodeling) and actomyosin contractility during vascular tubulogenesis. Rasip1 promotes activity of Cdc42 to activate Pak4, which in turn activates NMII, clearing apical junctions. Once lumens open, Rasip1 suppresses actomyosin contractility via inhibition of RhoA by Arhgap29, allowing controlled expansion of vessel lumens during embryonic growth. These findings elucidate the stepwise processes regulated by Rasip1 through downstream Rho GTPases and NMII.
Keywords: Ras homologue gene family member A GTP-binding protein; Ras interacting protein 1; actin cytoskeleton; endothelial cells; morphogenesis; myosin type II; tubulogenesis.
© 2016 American Heart Association, Inc.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
