

The Double-Stranded DNA Virosphere as a Modular Hierarchical Network of Gene Sharing
- PMID: 27486193
- PMCID: PMC4981718
- DOI: 10.1128/mBio.00978-16
The Double-Stranded DNA Virosphere as a Modular Hierarchical Network of Gene Sharing
Abstract
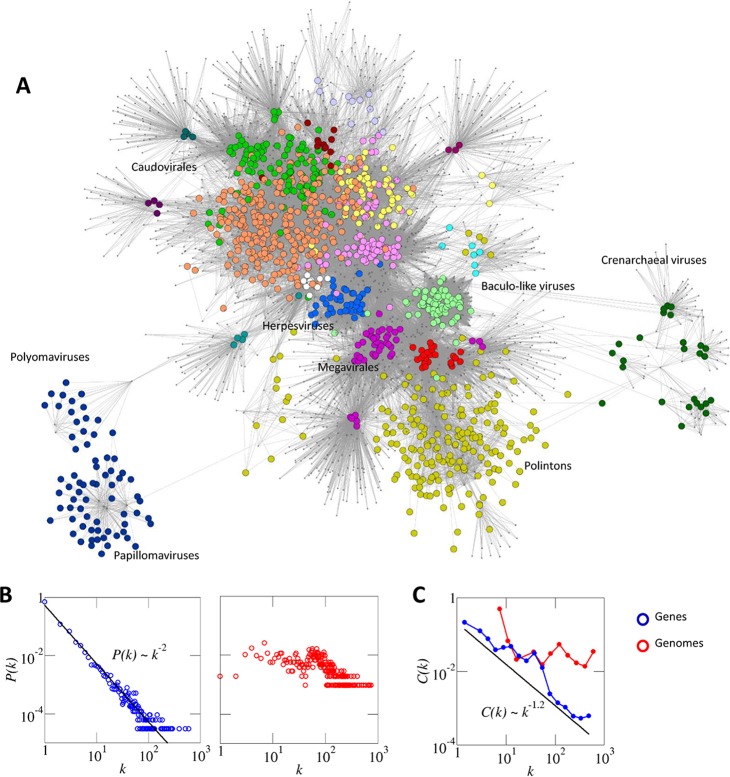
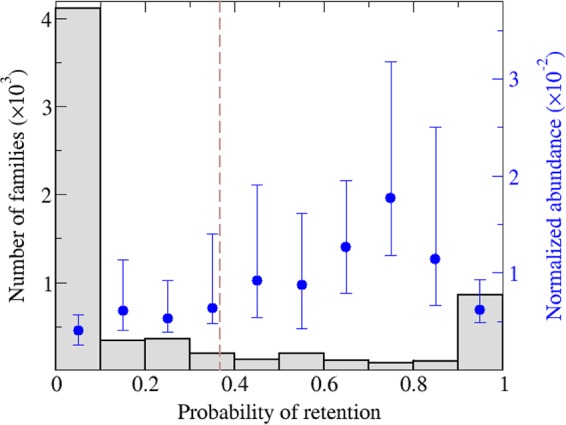
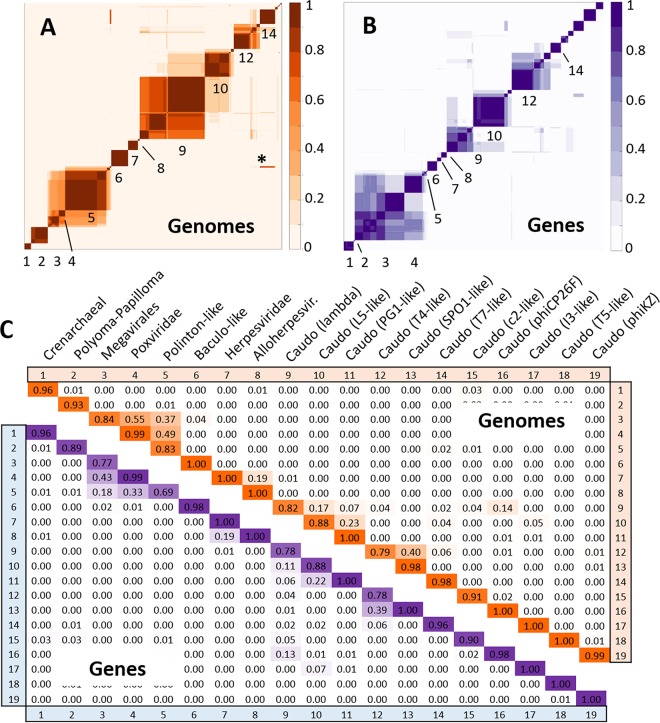
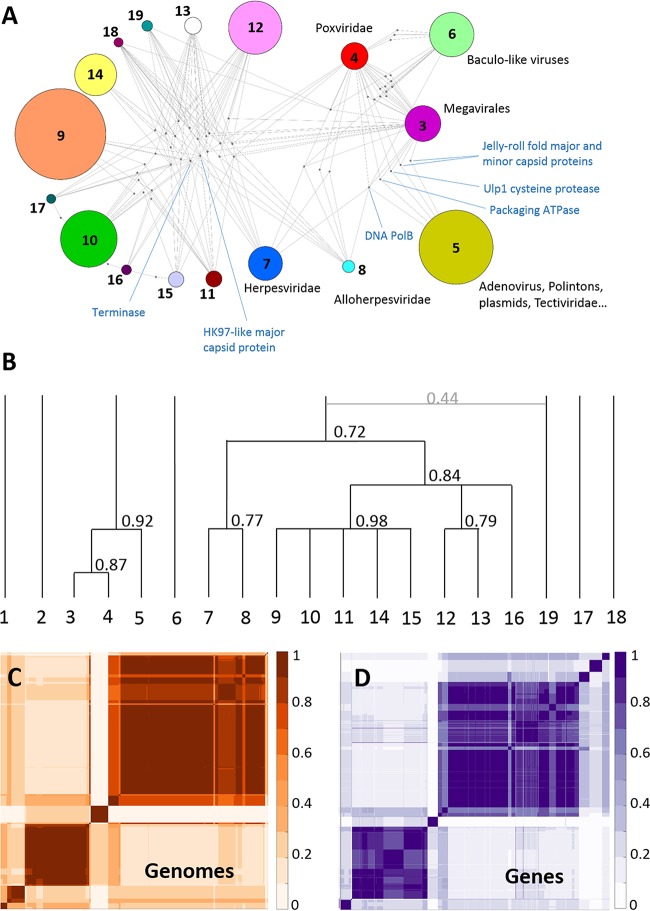
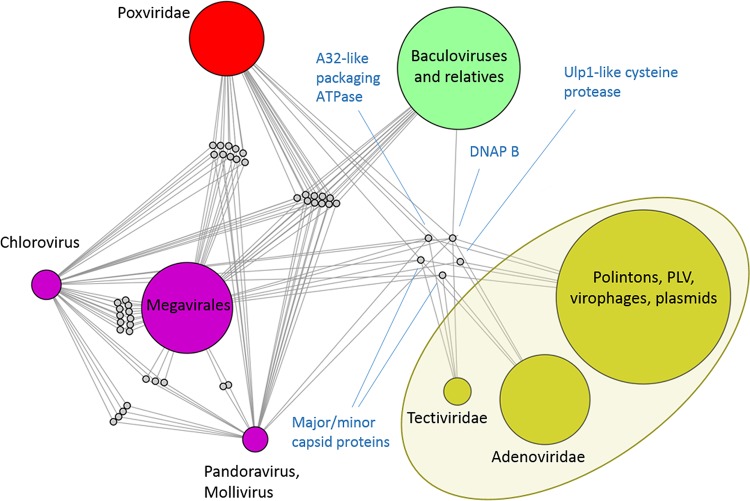
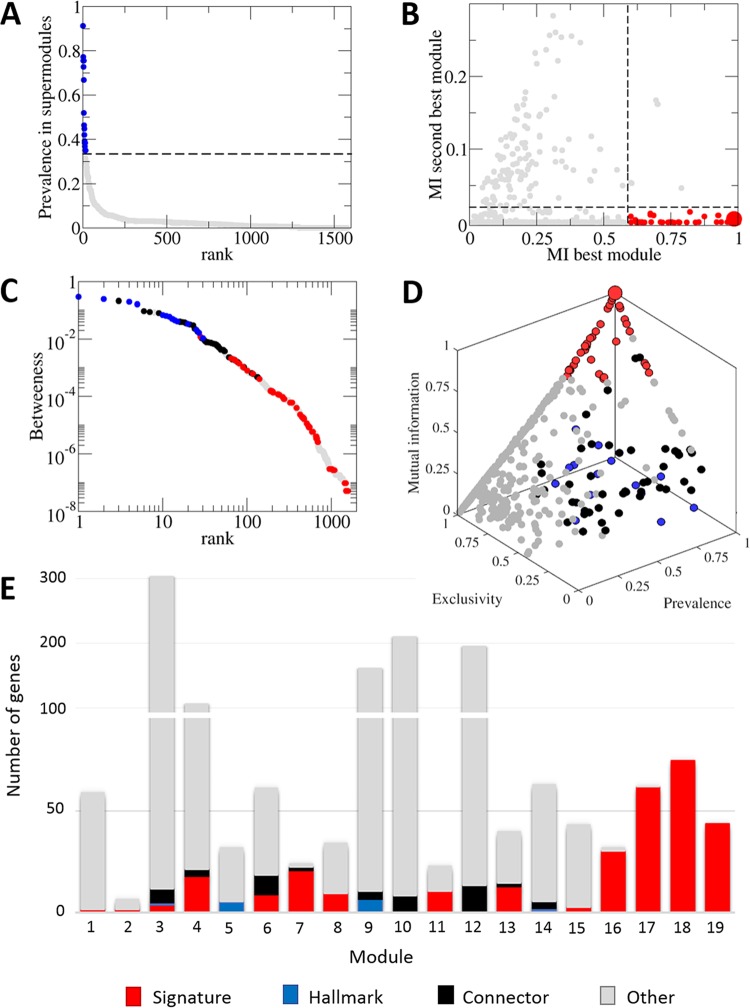
Virus genomes are prone to extensive gene loss, gain, and exchange and share no universal genes. Therefore, in a broad-scale study of virus evolution, gene and genome network analyses can complement traditional phylogenetics. We performed an exhaustive comparative analysis of the genomes of double-stranded DNA (dsDNA) viruses by using the bipartite network approach and found a robust hierarchical modularity in the dsDNA virosphere. Bipartite networks consist of two classes of nodes, with nodes in one class, in this case genomes, being connected via nodes of the second class, in this case genes. Such a network can be partitioned into modules that combine nodes from both classes. The bipartite network of dsDNA viruses includes 19 modules that form 5 major and 3 minor supermodules. Of these modules, 11 include tailed bacteriophages, reflecting the diversity of this largest group of viruses. The module analysis quantitatively validates and refines previously proposed nontrivial evolutionary relationships. An expansive supermodule combines the large and giant viruses of the putative order "Megavirales" with diverse moderate-sized viruses and related mobile elements. All viruses in this supermodule share a distinct morphogenetic tool kit with a double jelly roll major capsid protein. Herpesviruses and tailed bacteriophages comprise another supermodule, held together by a distinct set of morphogenetic proteins centered on the HK97-like major capsid protein. Together, these two supermodules cover the great majority of currently known dsDNA viruses. We formally identify a set of 14 viral hallmark genes that comprise the hubs of the network and account for most of the intermodule connections.
Importance: Viruses and related mobile genetic elements are the dominant biological entities on earth, but their evolution is not sufficiently understood and their classification is not adequately developed. The key reason is the characteristic high rate of virus evolution that involves not only sequence change but also extensive gene loss, gain, and exchange. Therefore, in the study of virus evolution on a large scale, traditional phylogenetic approaches have limited applicability and have to be complemented by gene and genome network analyses. We applied state-of-the art methods of such analysis to reveal robust hierarchical modularity in the genomes of double-stranded DNA viruses. Some of the identified modules combine highly diverse viruses infecting bacteria, archaea, and eukaryotes, in support of previous hypotheses on direct evolutionary relationships between viruses from the three domains of cellular life. We formally identify a set of 14 viral hallmark genes that hold together the genomic network.
Copyright © 2016 Iranzo et al.
Figures

Comment in
-
A network perspective on the virus world.Commun Integr Biol. 2017 Feb 23;10(2):e1296614. doi: 10.1080/19420889.2017.1296614. eCollection 2017. Commun Integr Biol. 2017. PMID: 28451057 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
