

Erasure of DNA methylation, genomic imprints, and epimutations in a primordial germ-cell model derived from mouse pluripotent stem cells
- PMID: 27486249
- PMCID: PMC5003264
- DOI: 10.1073/pnas.1610259113
Erasure of DNA methylation, genomic imprints, and epimutations in a primordial germ-cell model derived from mouse pluripotent stem cells
Erratum in
-
Correction for Miyoshi et al., Erasure of DNA methylation, genomic imprints, and epimutations in a primordial germ-cell model derived from mouse pluripotent stem cells.Proc Natl Acad Sci U S A. 2016 Sep 20;113(38):E5697. doi: 10.1073/pnas.1613505113. Epub 2016 Sep 12. Proc Natl Acad Sci U S A. 2016. PMID: 27621465 Free PMC article. No abstract available.
Abstract
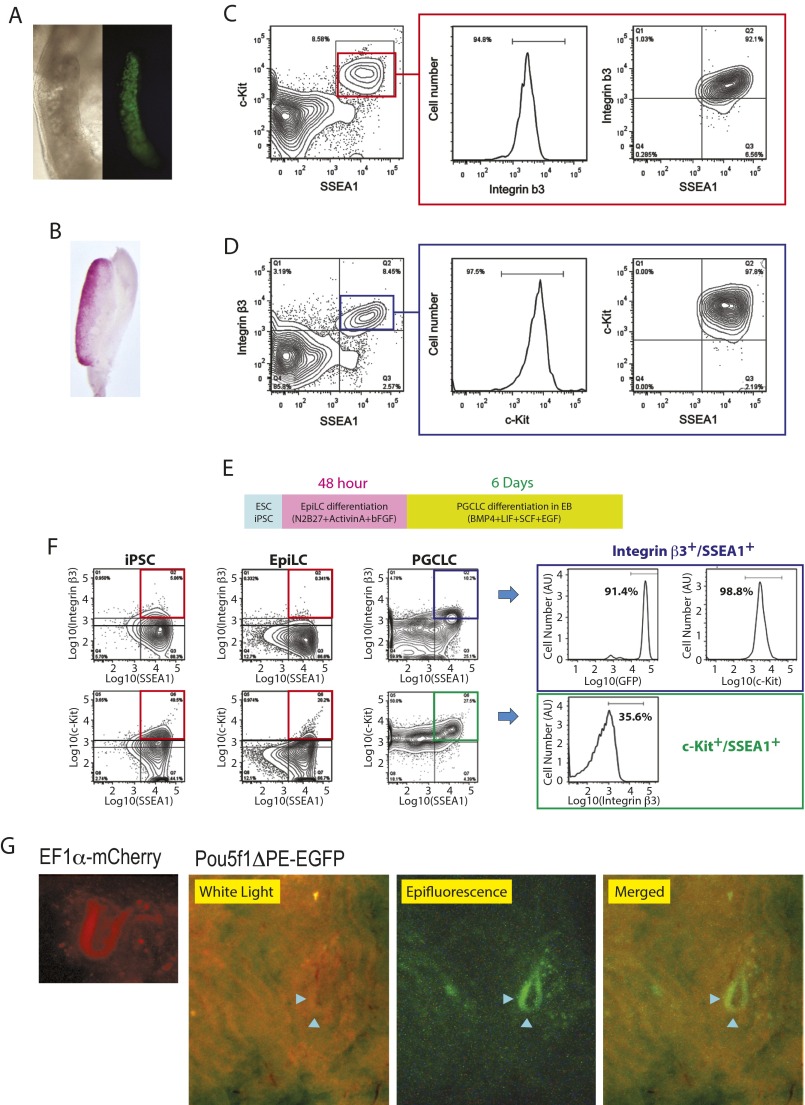
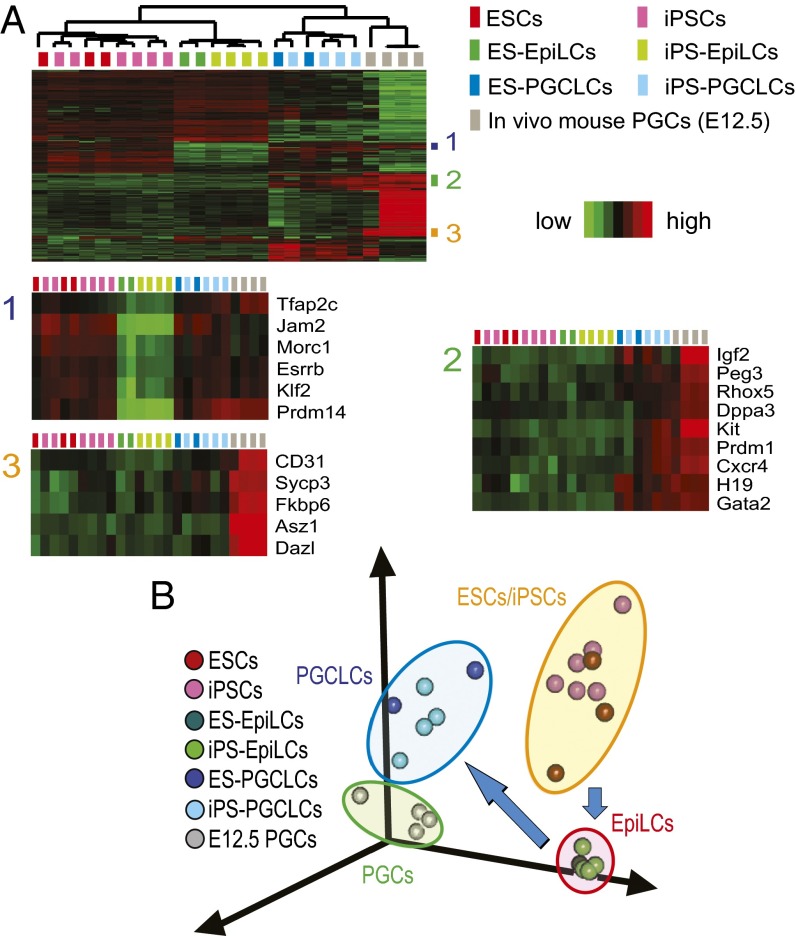
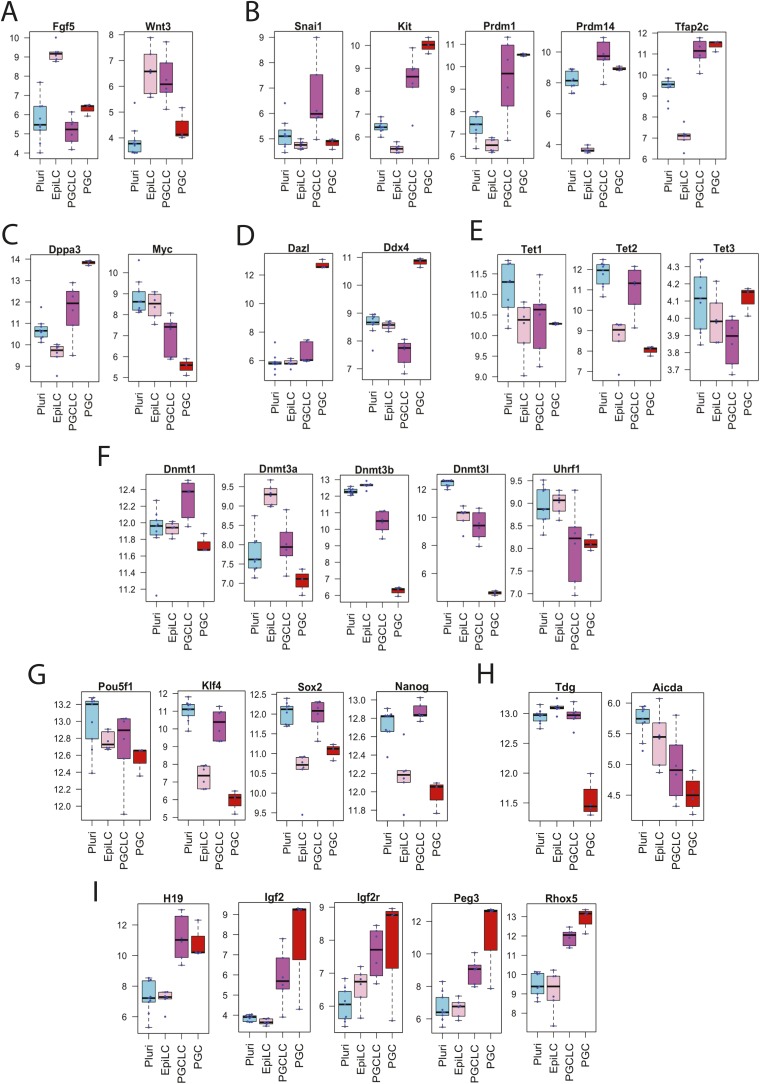
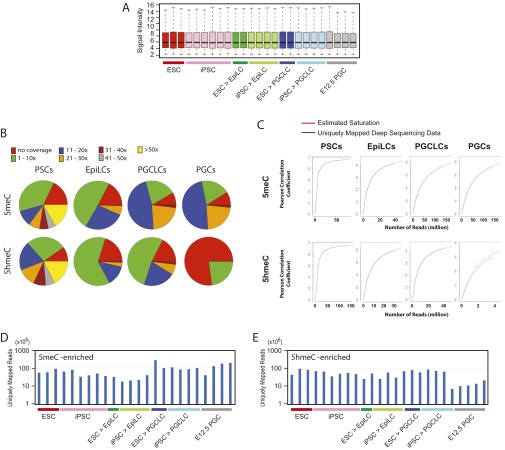
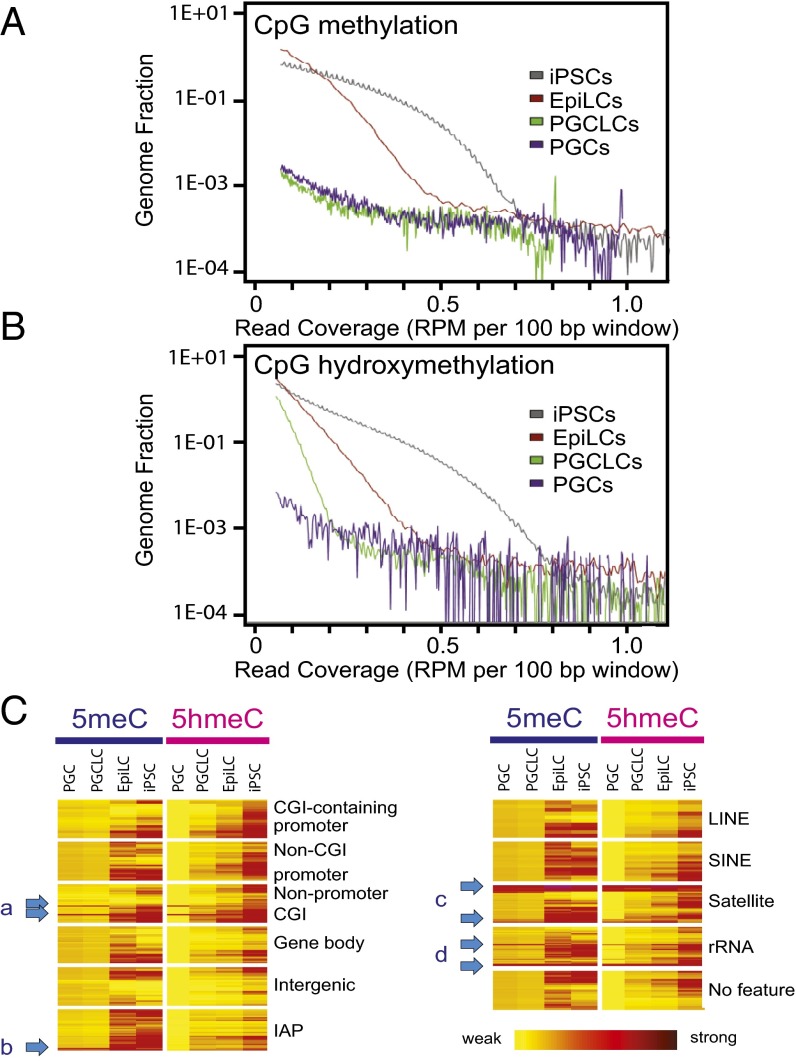
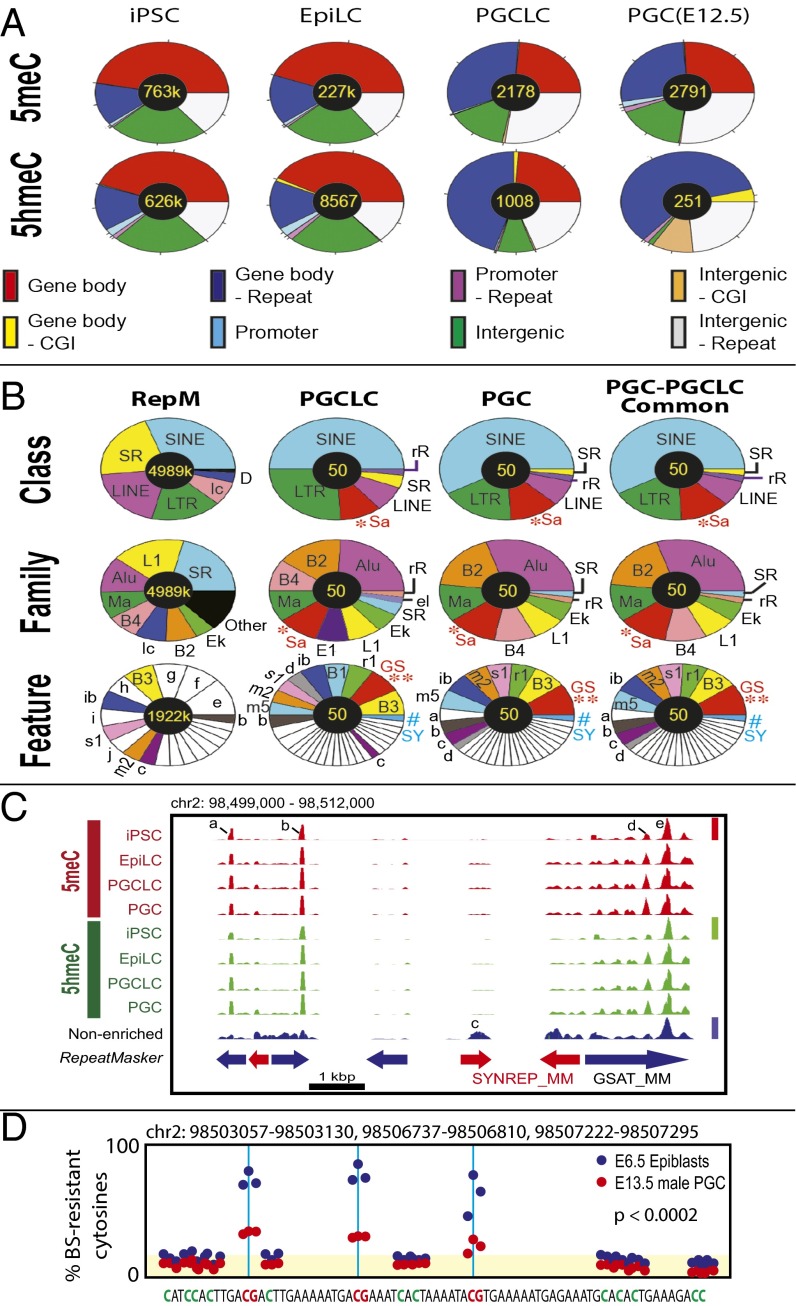
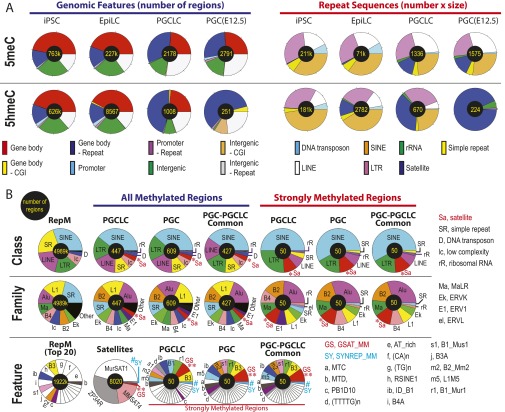
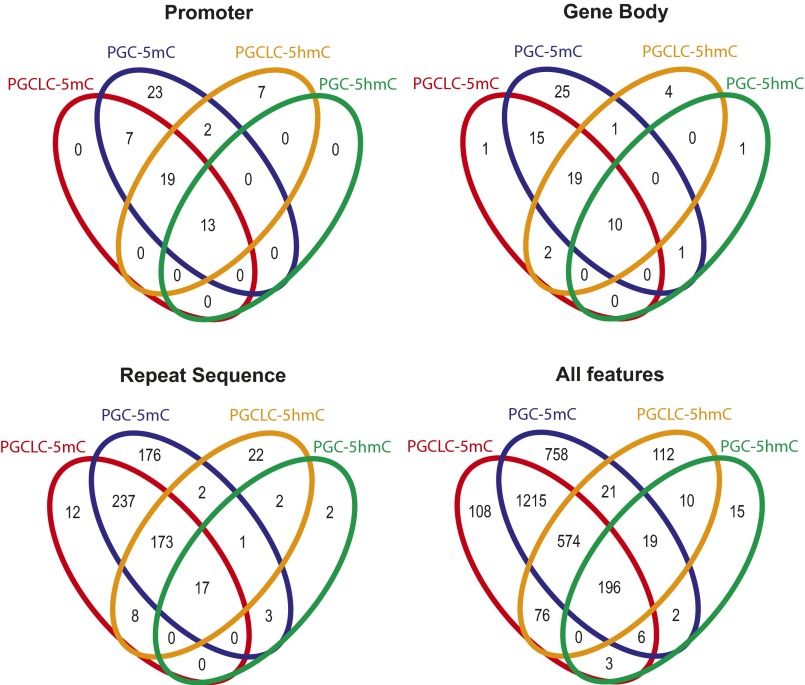
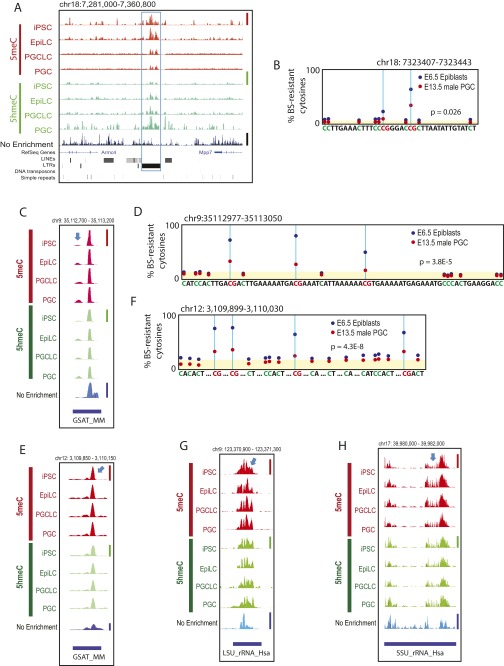
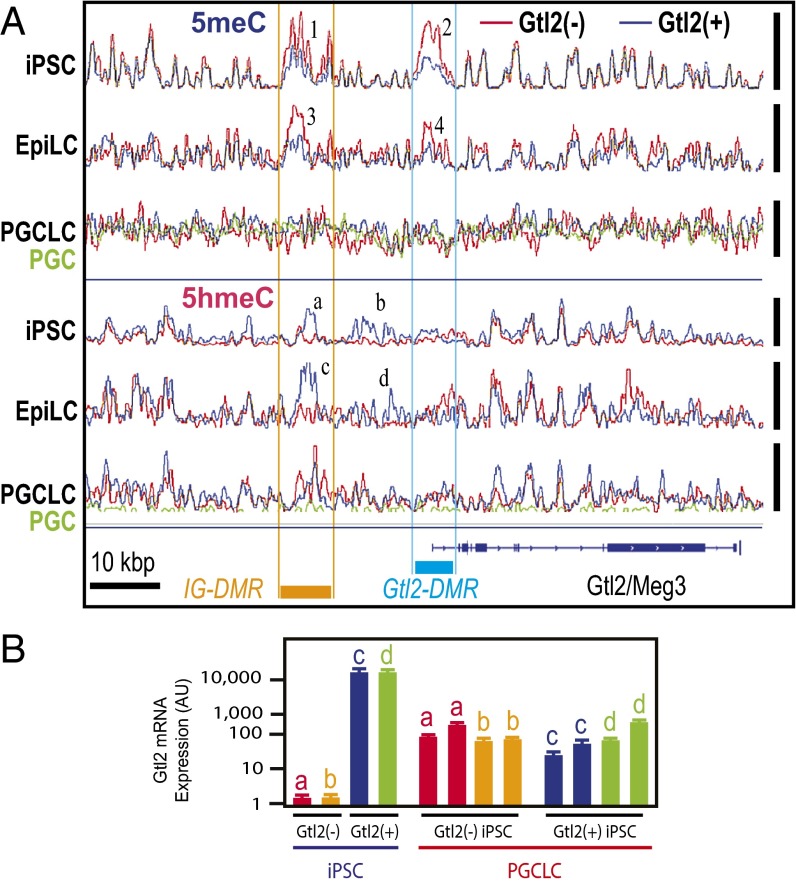
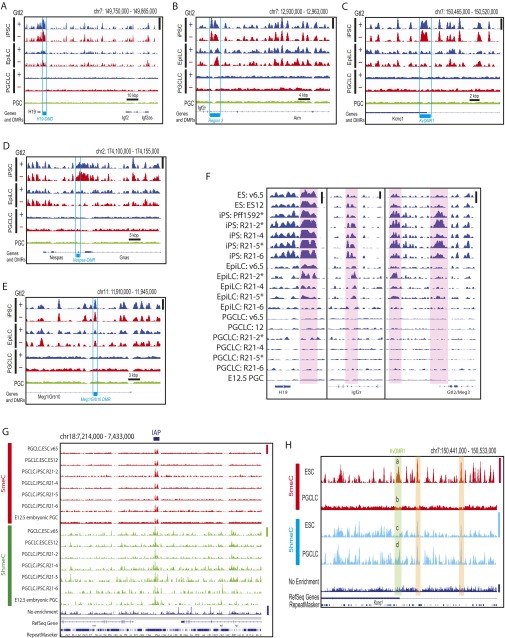
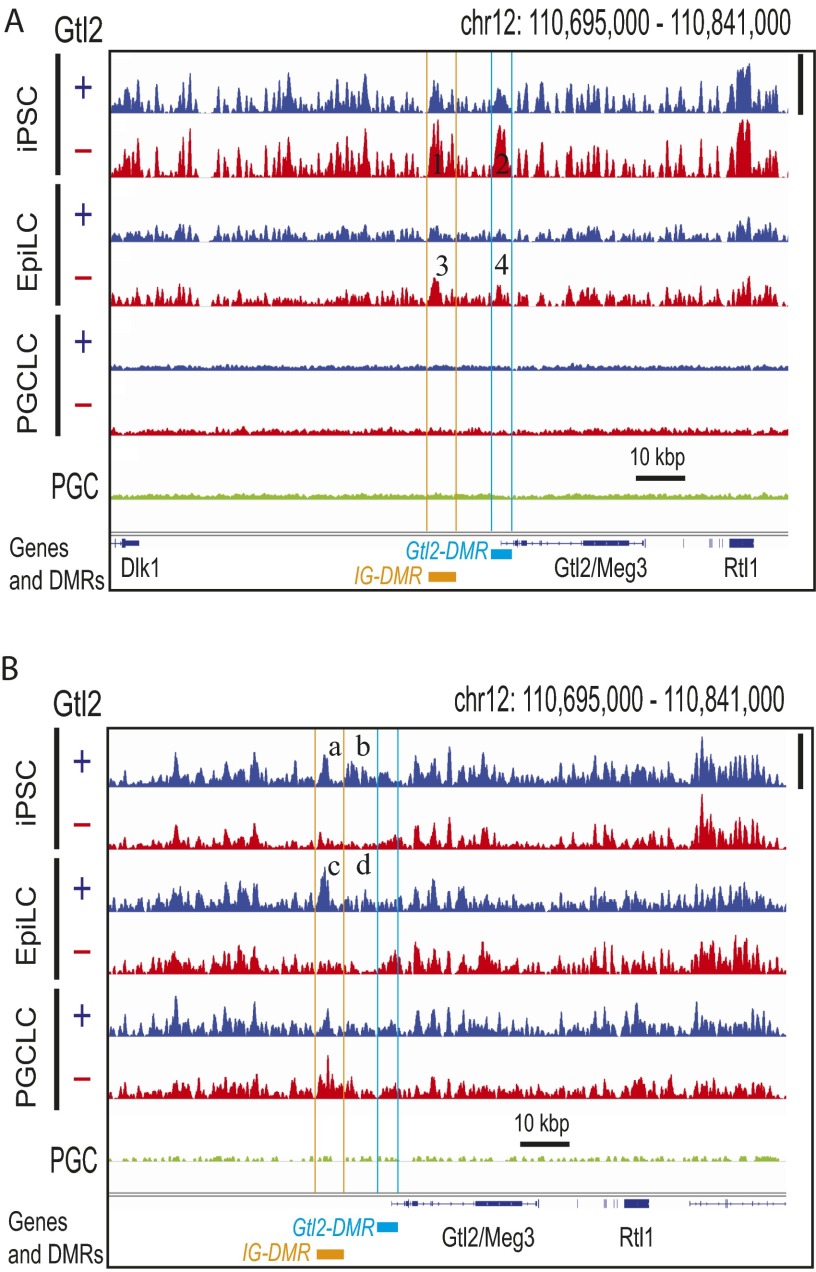
The genome-wide depletion of 5-methylcytosines (5meCs) caused by passive dilution through DNA synthesis without daughter strand methylation and active enzymatic processes resulting in replacement of 5meCs with unmethylated cytosines is a hallmark of primordial germ cells (PGCs). Although recent studies have shown that in vitro differentiation of pluripotent stem cells (PSCs) to PGC-like cells (PGCLCs) mimics the in vivo differentiation of epiblast cells to PGCs, how DNA methylation status of PGCLCs resembles the dynamics of 5meC erasure in embryonic PGCs remains controversial. Here, by differential detection of genome-wide 5meC and 5-hydroxymethylcytosine (5hmeC) distributions by deep sequencing, we show that PGCLCs derived from mouse PSCs recapitulated the process of genome-wide DNA demethylation in embryonic PGCs, including significant demethylation of imprint control regions (ICRs) associated with increased mRNA expression of the corresponding imprinted genes. Although 5hmeCs were also significantly diminished in PGCLCs, they retained greater amounts of 5hmeCs than intragonadal PGCs. The genomes of both PGCLCs and PGCs selectively retained both 5meCs and 5hmeCs at a small number of repeat sequences such as GSAT_MM, of which the significant retention of bisulfite-resistant cytosines was corroborated by reanalysis of previously published whole-genome bisulfite sequencing data for intragonadal PGCs. PSCs harboring abnormal hypermethylation at ICRs of the Dlk1-Gtl2-Dio3 imprinting cluster diminished these 5meCs upon differentiation to PGCLCs, resulting in transcriptional reactivation of the Gtl2 gene. These observations support the usefulness of PGCLCs in studying the germline epigenetic erasure including imprinted genes, epimutations, and erasure-resistant loci, which may be involved in transgenerational epigenetic inheritance.
Keywords: PGCLC; epigenetic reprogramming; epimutation; genetic imprinting.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency.Nature. 2010 Feb 25;463(7284):1101-5. doi: 10.1038/nature08829. Nature. 2010. PMID: 20098412 Free PMC article.
-
Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine.Science. 2013 Jan 25;339(6118):448-52. doi: 10.1126/science.1229277. Epub 2012 Dec 6. Science. 2013. PMID: 23223451 Free PMC article.
-
Active DNA demethylation is required for complete imprint erasure in primordial germ cells.Sci Rep. 2014 Jan 13;4:3658. doi: 10.1038/srep03658. Sci Rep. 2014. PMID: 24413819 Free PMC article.
-
A current view of the epigenome in mouse primordial germ cells.Mol Reprod Dev. 2014 Feb;81(2):160-70. doi: 10.1002/mrd.22214. Epub 2013 Aug 7. Mol Reprod Dev. 2014. PMID: 23868517 Review.
-
Epigenome regulation during germ cell specification and development from pluripotent stem cells.Curr Opin Genet Dev. 2018 Oct;52:57-64. doi: 10.1016/j.gde.2018.06.004. Epub 2018 Jun 13. Curr Opin Genet Dev. 2018. PMID: 29908427 Review.
Cited by
-
Generation of Human Primordial Germ Cell-like Cells at the Surface of Embryoid Bodies from Primed-pluripotency Induced Pluripotent Stem Cells.J Vis Exp. 2019 Jan 11;(143):10.3791/58297. doi: 10.3791/58297. J Vis Exp. 2019. PMID: 30688291 Free PMC article.
-
Germline genome protection: implications for gamete quality and germ cell tumorigenesis.Andrology. 2019 Jul;7(4):516-526. doi: 10.1111/andr.12651. Epub 2019 May 22. Andrology. 2019. PMID: 31119900 Free PMC article. Review.
-
Bend family proteins mark chromatin boundaries and synergistically promote early germ cell differentiation.Protein Cell. 2022 Oct;13(10):721-741. doi: 10.1007/s13238-021-00884-1. Epub 2021 Nov 3. Protein Cell. 2022. PMID: 34731408 Free PMC article.
-
Male obesity impacts DNA methylation reprogramming in sperm.Clin Epigenetics. 2021 Jan 25;13(1):17. doi: 10.1186/s13148-020-00997-0. Clin Epigenetics. 2021. PMID: 33494820 Free PMC article.
-
Integrated Analyses of Mouse Stem Cell Transcriptomes Provide Clues for Stem Cell Maintenance and Transdifferentiation.Front Genet. 2020 Sep 4;11:563798. doi: 10.3389/fgene.2020.563798. eCollection 2020. Front Genet. 2020. PMID: 33101382 Free PMC article.
References
-
- Prokopuk L, Western PS, Stringer JM. Transgenerational epigenetic inheritance: Adaptation through the germline epigenome? Epigenomics. 2015;7(5):829–846. - PubMed
-
- Szyf M. Nongenetic inheritance and transgenerational epigenetics. Trends Mol Med. 2015;21(2):134–144. - PubMed
-
- Bohacek J, Mansuy IM. Molecular insights into transgenerational non-genetic inheritance of acquired behaviours. Nat Rev Genet. 2015;16(11):641–652. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
