

Isolated and Syndromic Retinal Dystrophy Caused by Biallelic Mutations in RCBTB1, a Gene Implicated in Ubiquitination
- PMID: 27486781
- PMCID: PMC4974088
- DOI: 10.1016/j.ajhg.2016.06.017
Isolated and Syndromic Retinal Dystrophy Caused by Biallelic Mutations in RCBTB1, a Gene Implicated in Ubiquitination
Abstract
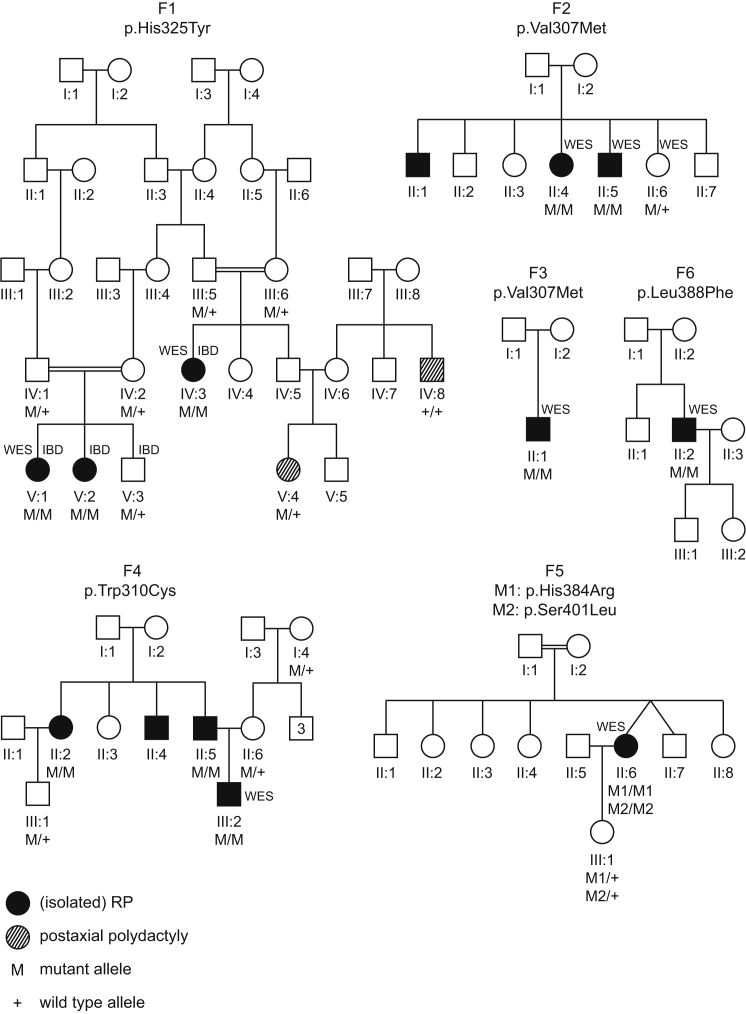
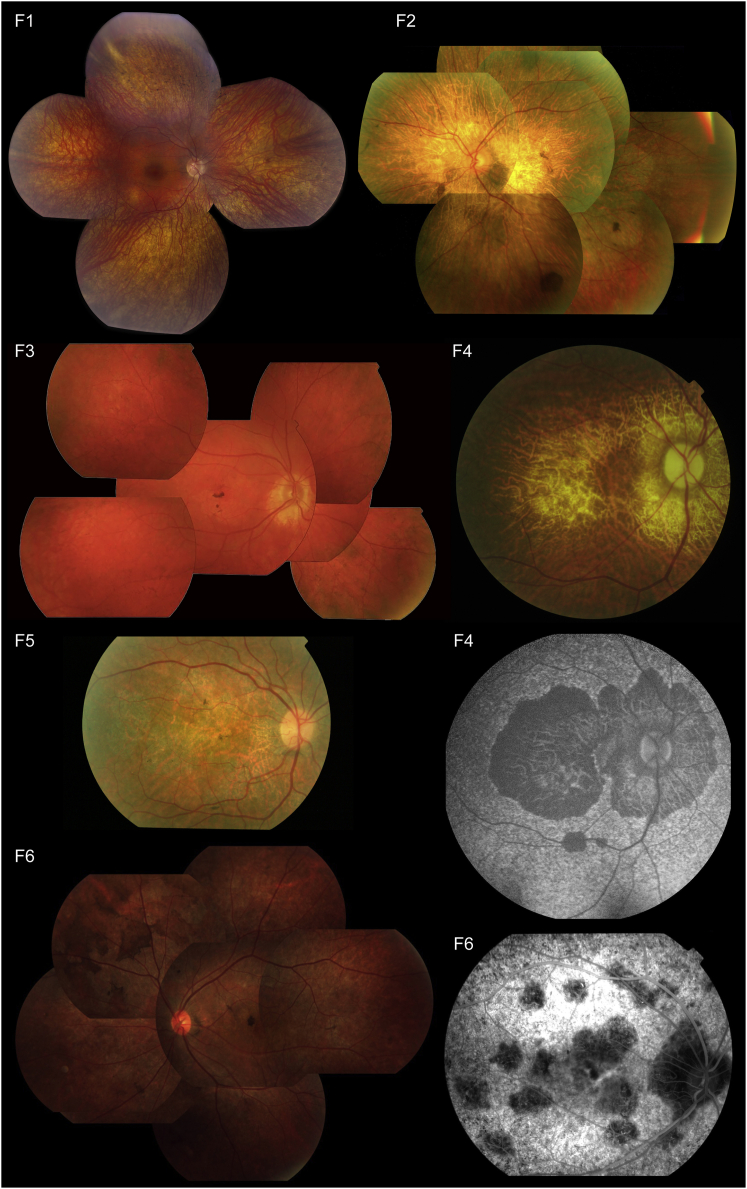
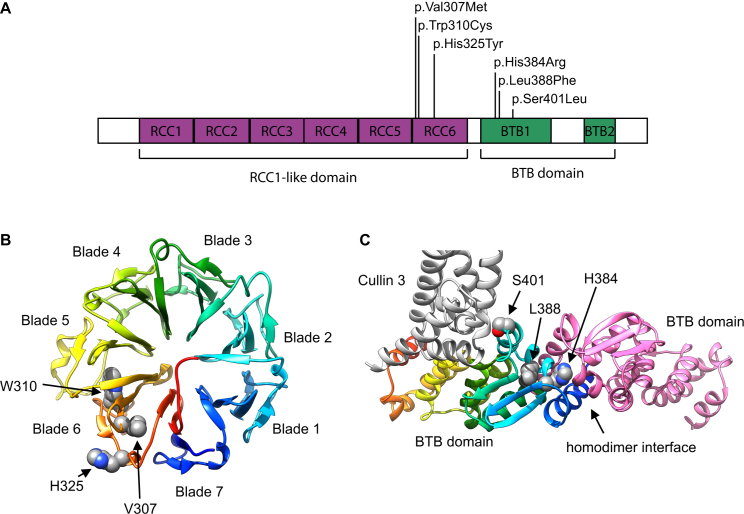
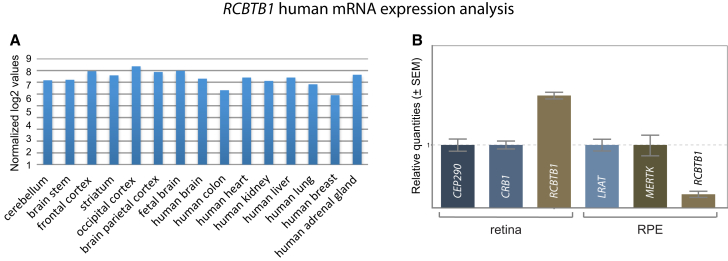
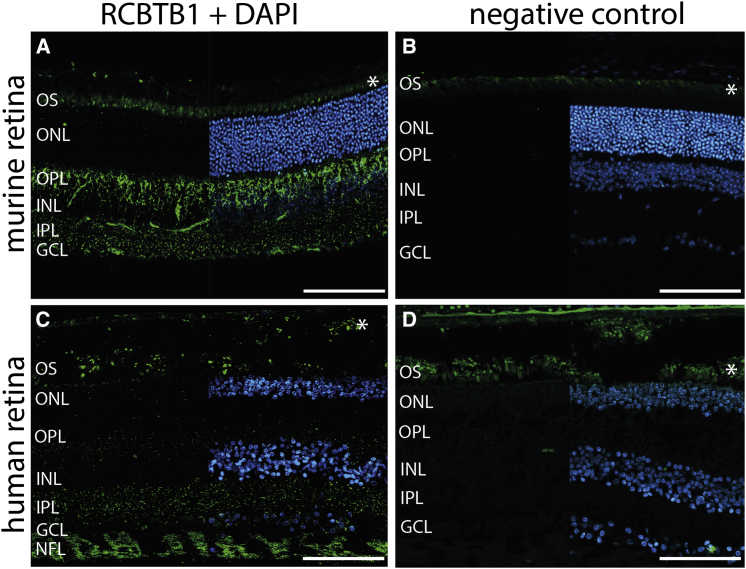
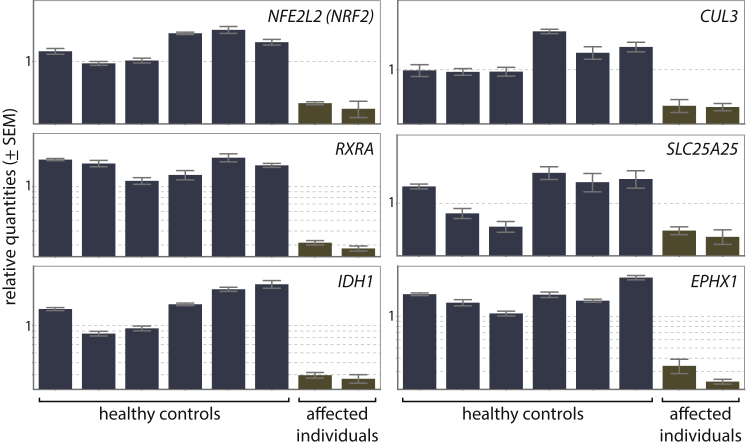
Inherited retinal dystrophies (iRDs) are a group of genetically and clinically heterogeneous conditions resulting from mutations in over 250 genes. Here, homozygosity mapping and whole-exome sequencing (WES) in a consanguineous family revealed a homozygous missense mutation, c.973C>T (p.His325Tyr), in RCBTB1. In affected individuals, it was found to segregate with retinitis pigmentosa (RP), goiter, primary ovarian insufficiency, and mild intellectual disability. Subsequent analysis of WES data in different cohorts uncovered four additional homozygous missense mutations in five unrelated families in whom iRD segregates with or without syndromic features. Ocular phenotypes ranged from typical RP starting in the second decade to chorioretinal dystrophy with a later age of onset. The five missense mutations affect highly conserved residues either in the sixth repeat of the RCC1 domain or in the BTB1 domain. A founder haplotype was identified for mutation c.919G>A (p.Val307Met), occurring in two families of Mediterranean origin. We showed ubiquitous mRNA expression of RCBTB1 and demonstrated predominant RCBTB1 localization in human inner retina. RCBTB1 was very recently shown to be involved in ubiquitination, more specifically as a CUL3 substrate adaptor. Therefore, the effect on different components of the CUL3 and NFE2L2 (NRF2) pathway was assessed in affected individuals' lymphocytes, revealing decreased mRNA expression of NFE2L2 and several NFE2L2 target genes. In conclusion, our study puts forward mutations in RCBTB1 as a cause of autosomal-recessive non-syndromic and syndromic iRD. Finally, our data support a role for impaired ubiquitination in the pathogenetic mechanism of RCBTB1 mutations.
Copyright © 2016 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Berger W., Kloeckener-Gruissem B., Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010;29:335–375. - PubMed
-
- Lee K., Garg S. Navigating the current landscape of clinical genetic testing for inherited retinal dystrophies. Genet. Med. 2015;17:245–252. - PubMed
-
- Lenassi E., Vincent A., Li Z., Saihan Z., Coffey A.J., Steele-Stallard H.B., Moore A.T., Steel K.P., Luxon L.M., Héon E. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 2015;23:1318–1327. - PMC - PubMed
-
- Roosing S., van den Born L.I., Sangermano R., Banfi S., Koenekoop R.K., Zonneveld-Vrieling M.N., Klaver C.C., van Lith-Verhoeven J.J., Cremers F.P., den Hollander A.I., Hoyng C.B. Mutations in MFSD8, encoding a lysosomal membrane protein, are associated with nonsyndromic autosomal recessive macular dystrophy. Ophthalmology. 2015;122:170–179. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
