

SAR11 bacteria linked to ocean anoxia and nitrogen loss
- PMID: 27487207
- PMCID: PMC4990128
- DOI: 10.1038/nature19068
SAR11 bacteria linked to ocean anoxia and nitrogen loss
Abstract
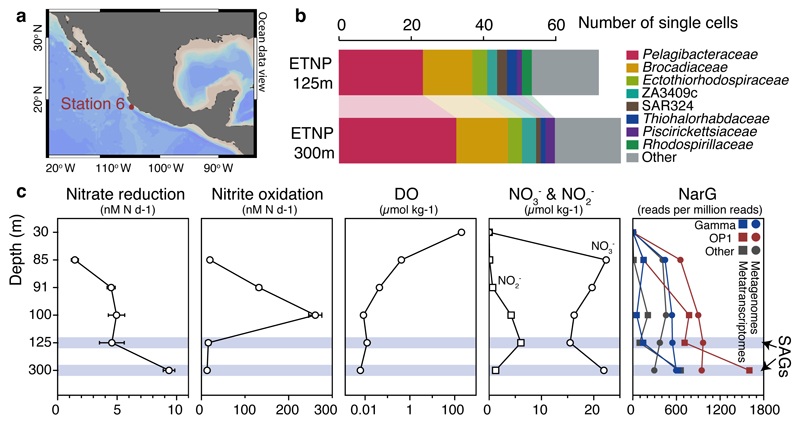
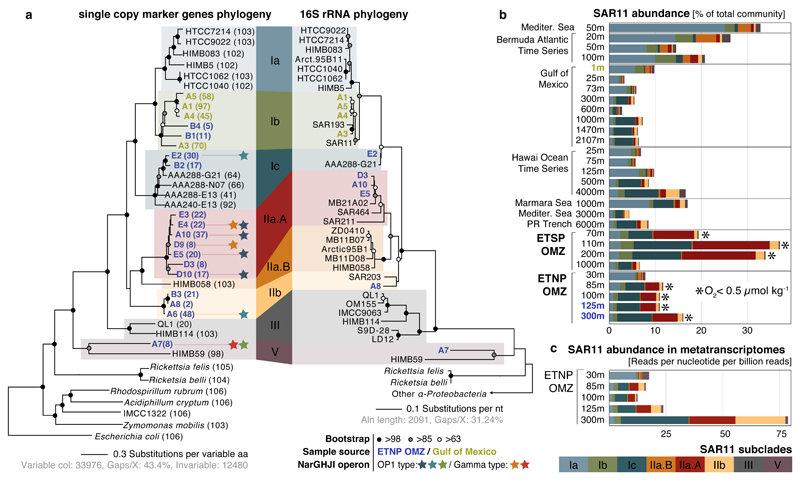
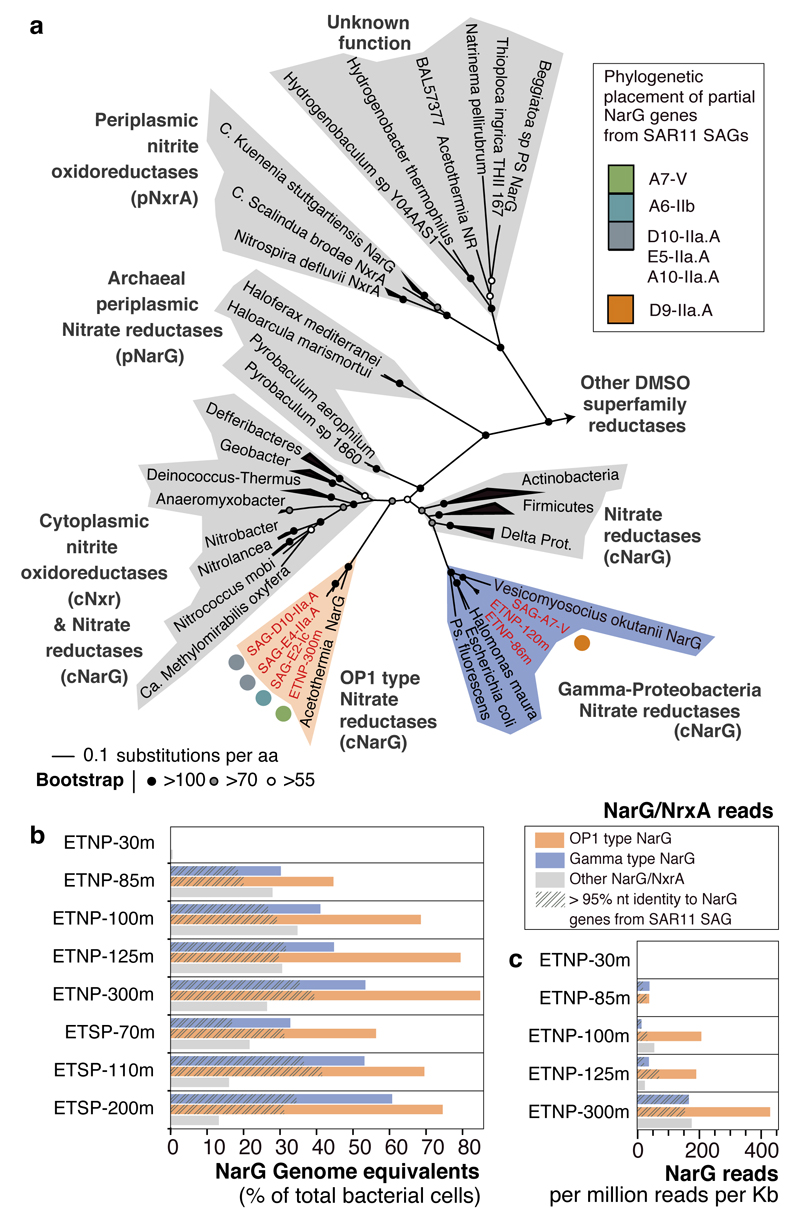
Bacteria of the SAR11 clade constitute up to one half of all microbial cells in the oxygen-rich surface ocean. SAR11 bacteria are also abundant in oxygen minimum zones (OMZs), where oxygen falls below detection and anaerobic microbes have vital roles in converting bioavailable nitrogen to N2 gas. Anaerobic metabolism has not yet been observed in SAR11, and it remains unknown how these bacteria contribute to OMZ biogeochemical cycling. Here, genomic analysis of single cells from the world's largest OMZ revealed previously uncharacterized SAR11 lineages with adaptations for life without oxygen, including genes for respiratory nitrate reductases (Nar). SAR11 nar genes were experimentally verified to encode proteins catalysing the nitrite-producing first step of denitrification and constituted ~40% of OMZ nar transcripts, with transcription peaking in the anoxic zone of maximum nitrate reduction activity. These results link SAR11 to pathways of ocean nitrogen loss, redefining the ecological niche of Earth's most abundant organismal group.
Conflict of interest statement
The authors declare no competing financial interests in association with this study.
Figures
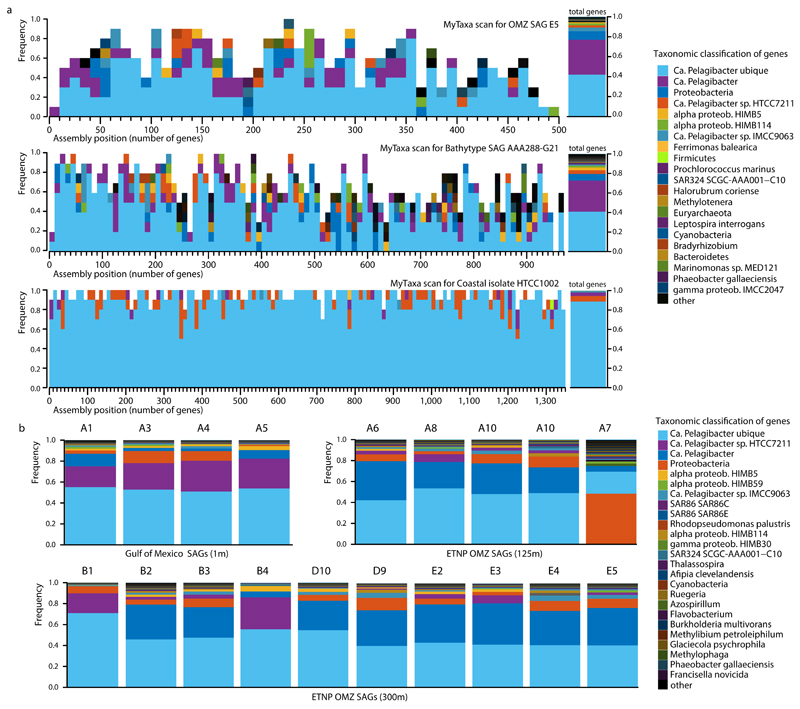
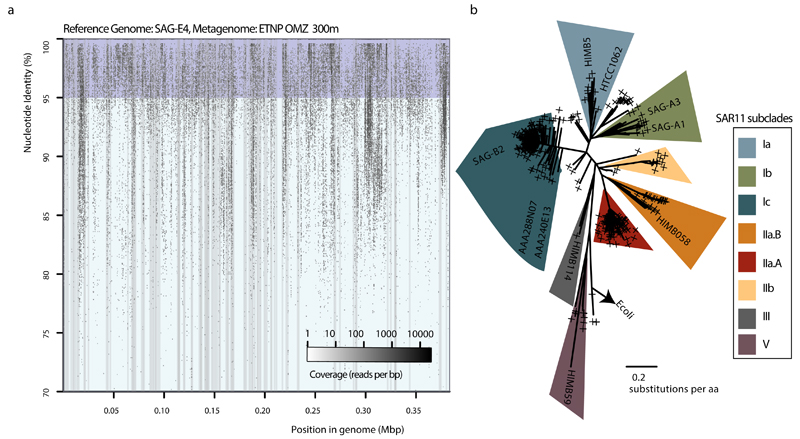
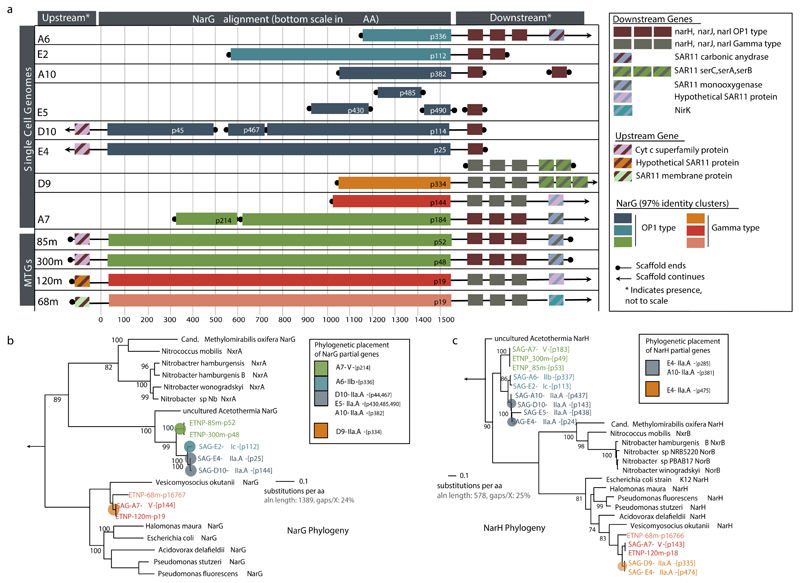
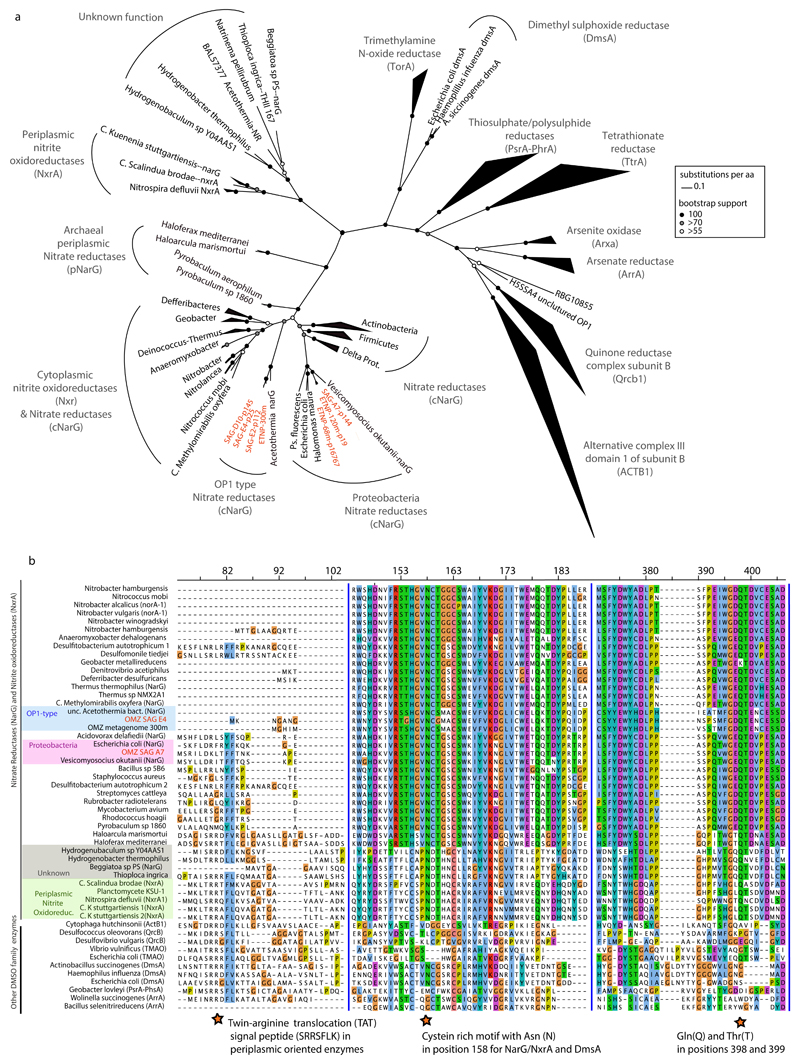
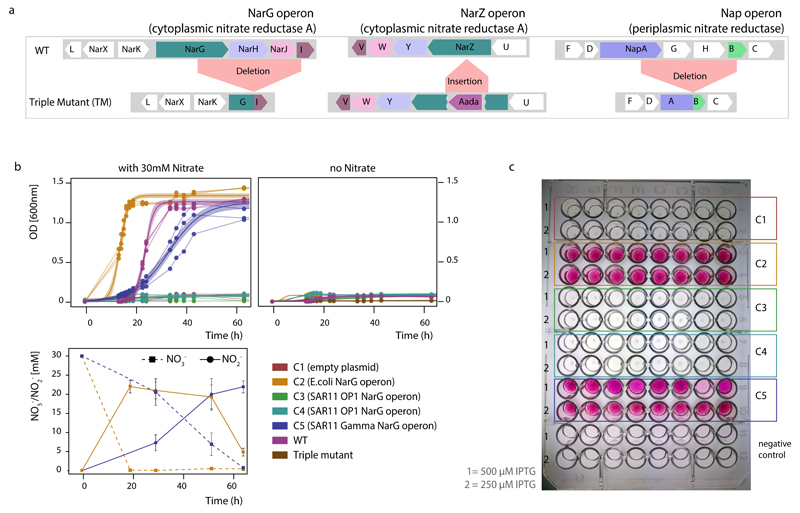
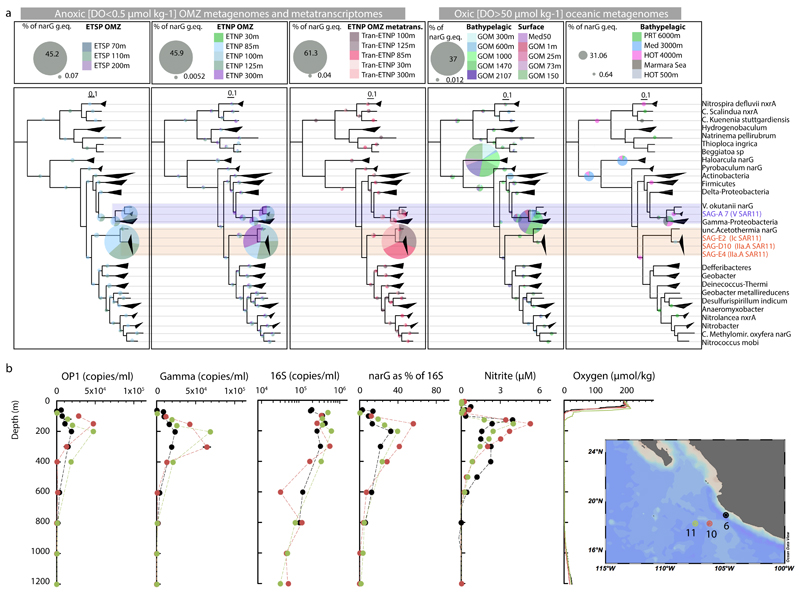
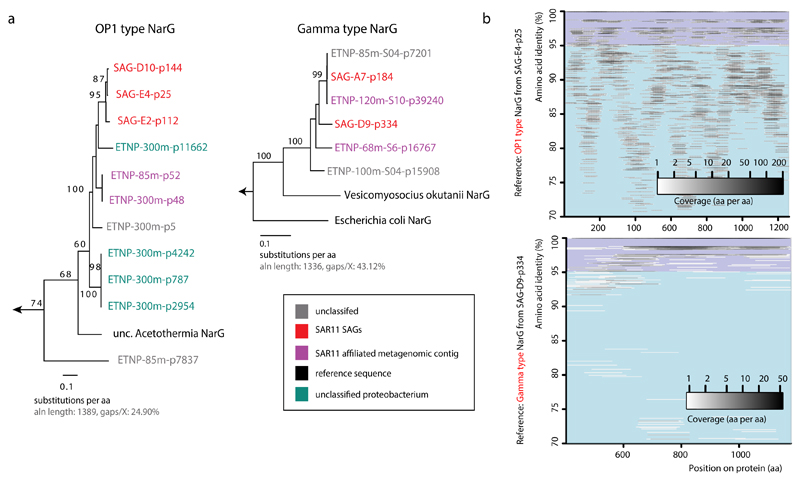
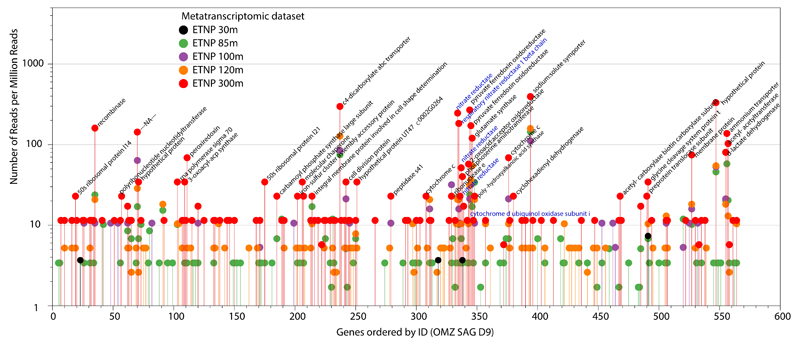
References
-
- Brown MV, Schwalbach MS, Hewson I, Fuhrman JA. Coupling 16S-ITS rDNA clone libraries and automated ribosomal intergenic spacer analysis to show marine microbial diversity: development and application to a time series. Environ Microbiol. 2005;7:1466–1479. - PubMed
-
- Carlson CA, et al. Seasonal dynamics of SAR11 populations in the euphotic and mesopelagic zones of the northwestern Sargasso Sea. ISME J. 2008;3:283–295. - PubMed
-
- Eiler A, Hayakawa DH, Church MJ, Karl DM, Rappé MS. Dynamics of the SAR11 bacterioplankton lineage in relation to environmental conditions in the oligotrophic North Pacific subtropical gyre. Environ Microbiol. 2009;11:2291–2300. - PubMed
-
- Morris RM, et al. SAR11 clade dominates ocean surface bacterioplankton communities. Nature. 2002;420:806–810. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
