

Emergence of a Homo sapiens-specific gene family and chromosome 16p11.2 CNV susceptibility
- PMID: 27487209
- PMCID: PMC4988886
- DOI: 10.1038/nature19075
Emergence of a Homo sapiens-specific gene family and chromosome 16p11.2 CNV susceptibility
Abstract
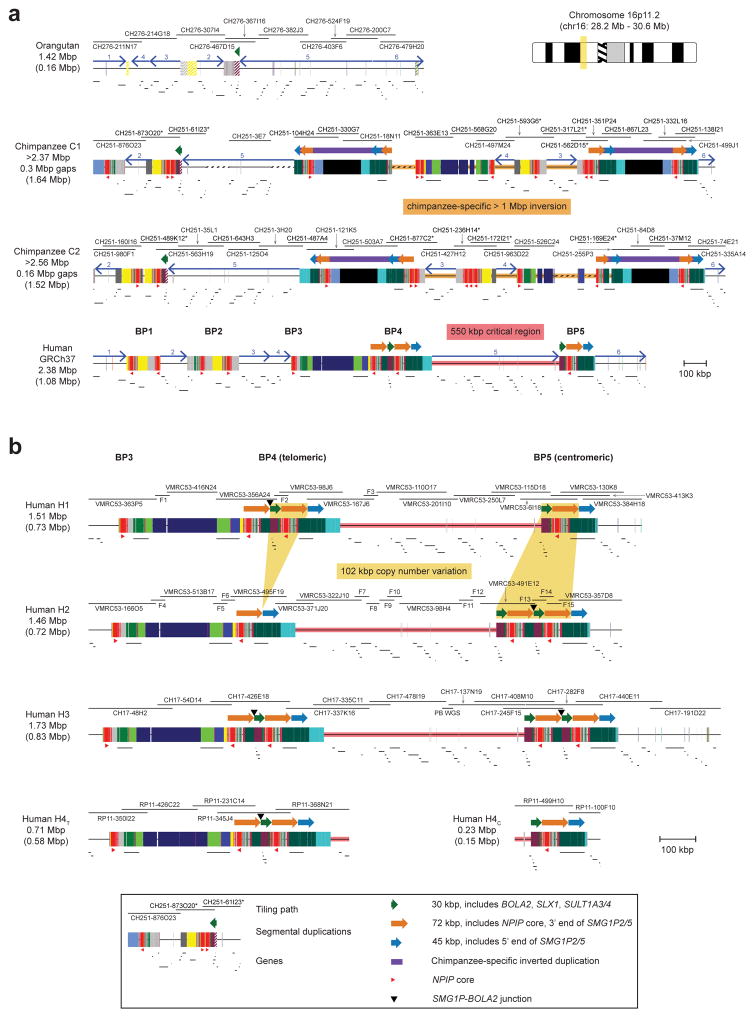
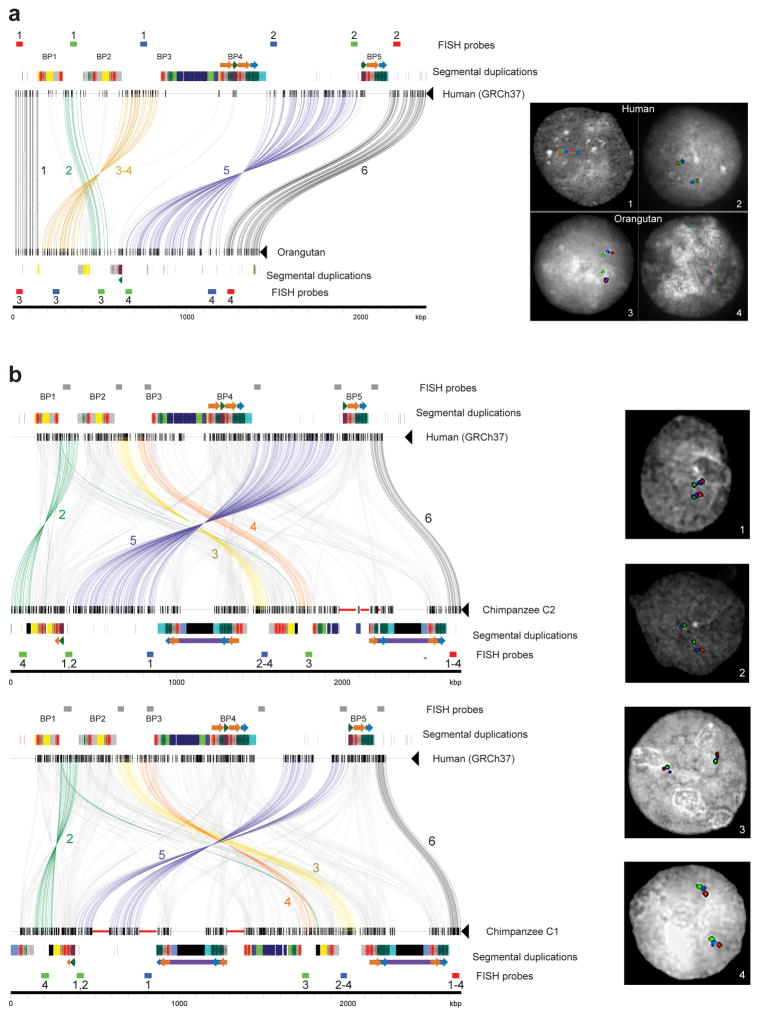
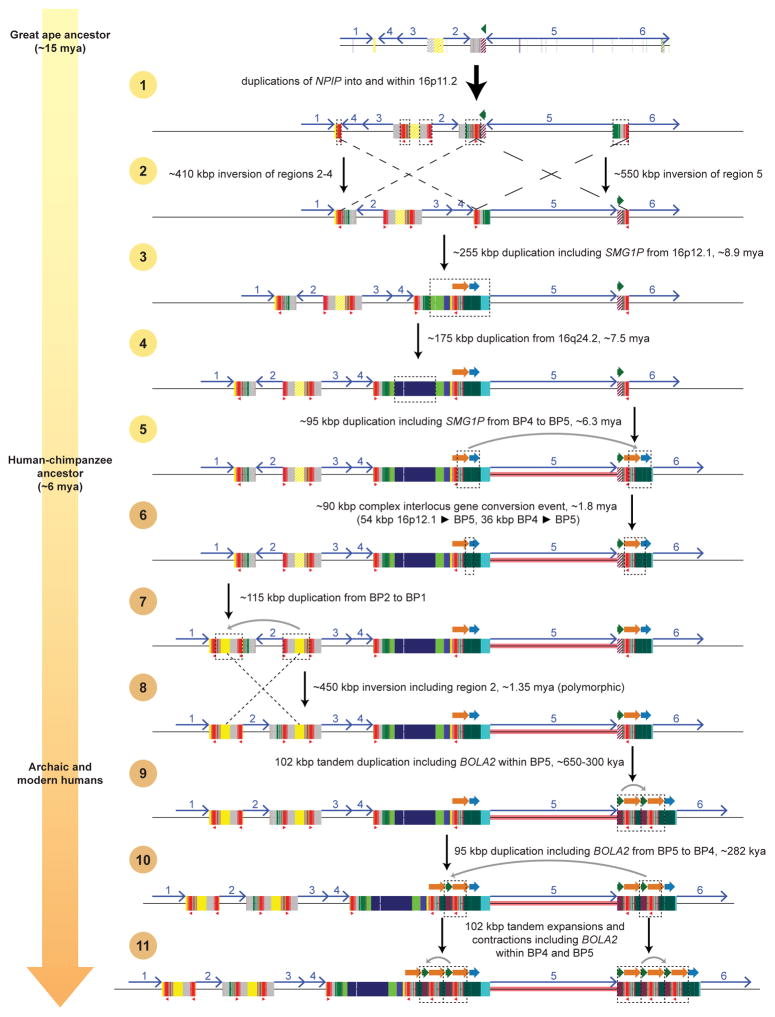
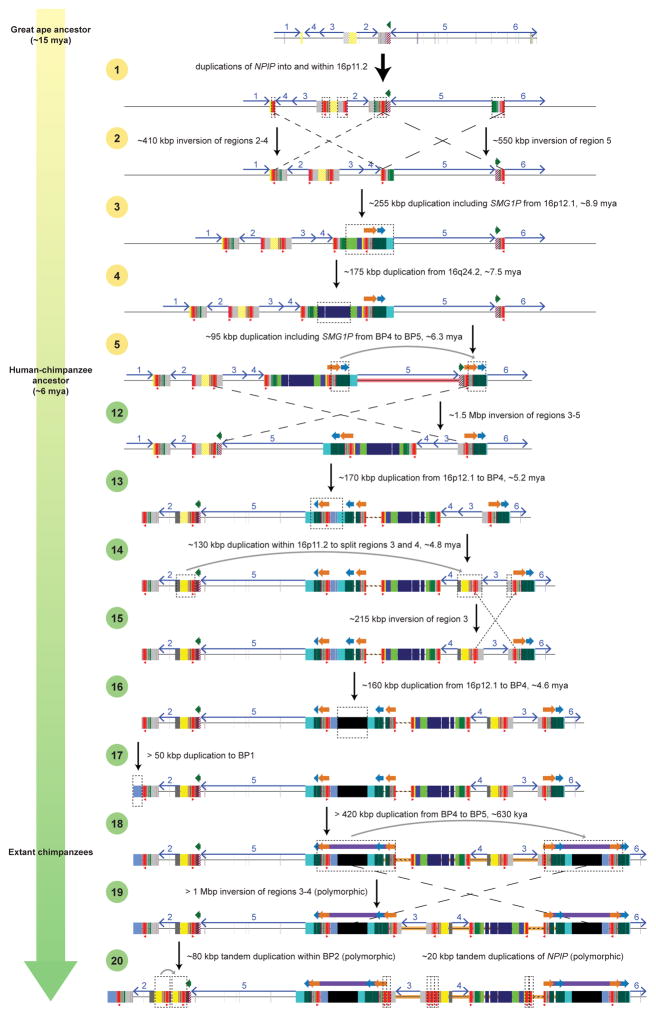
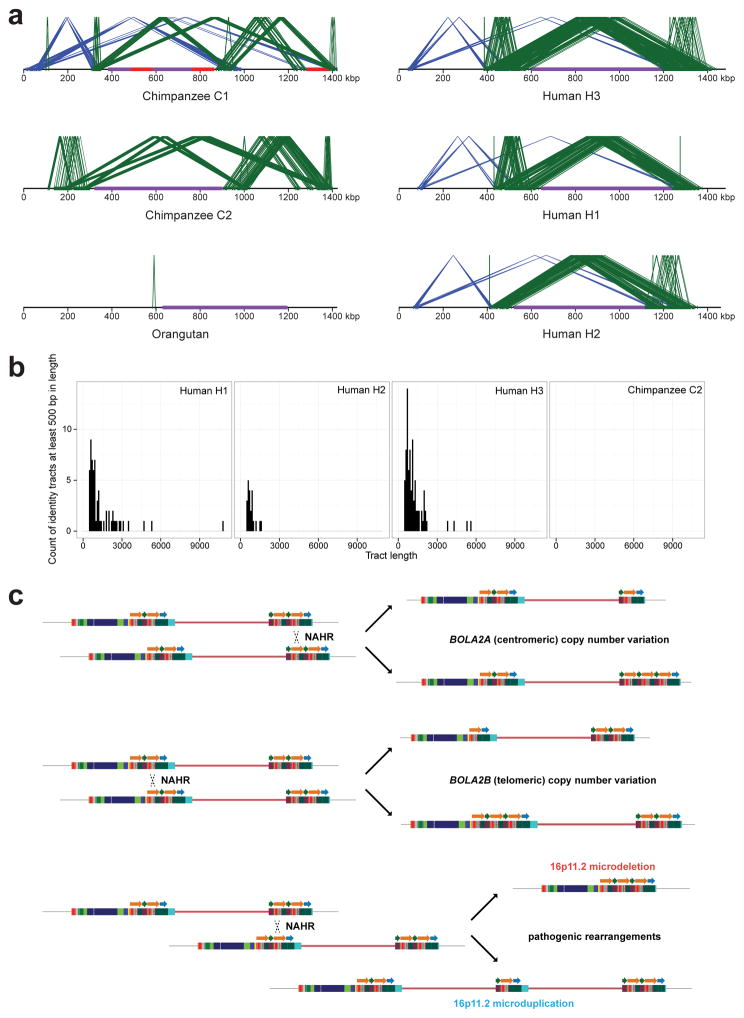
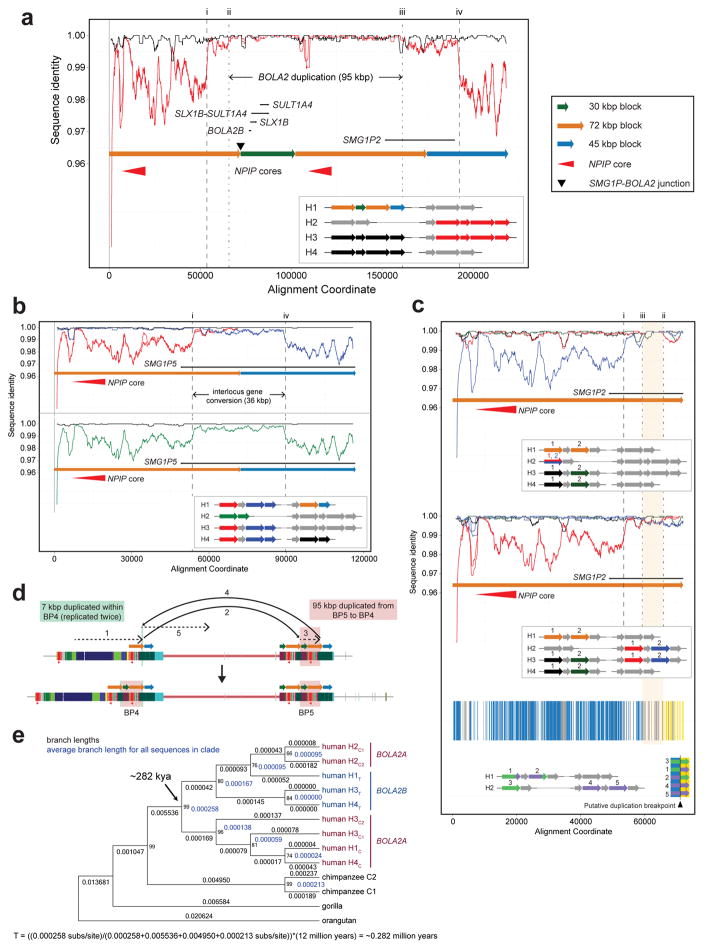
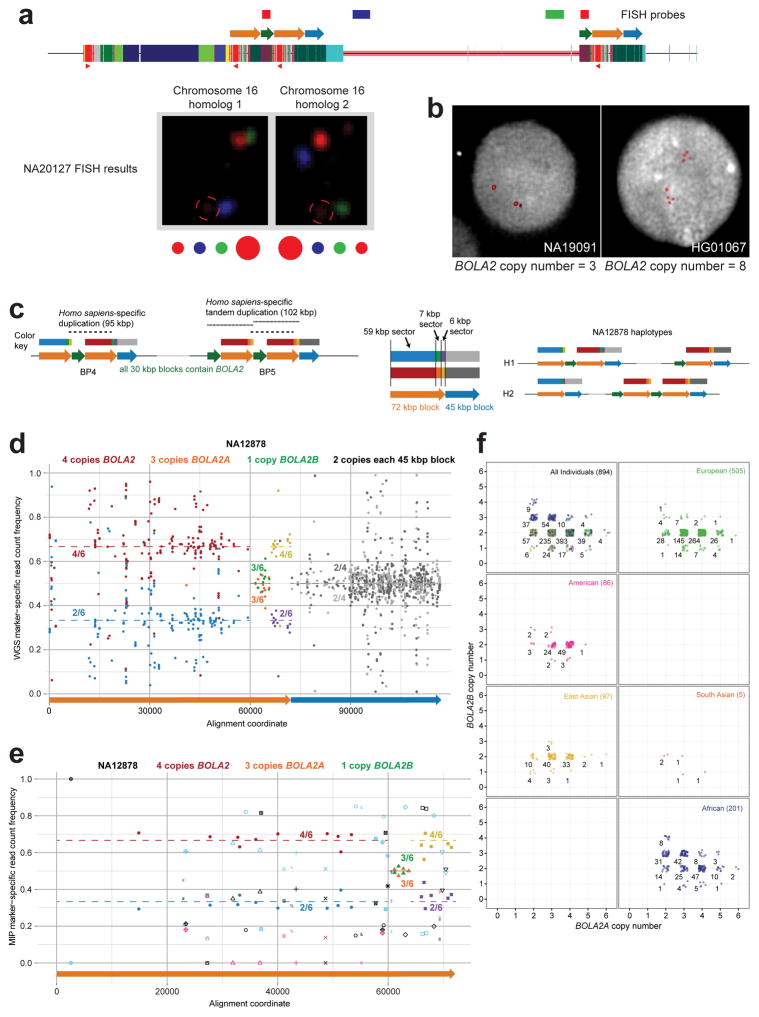
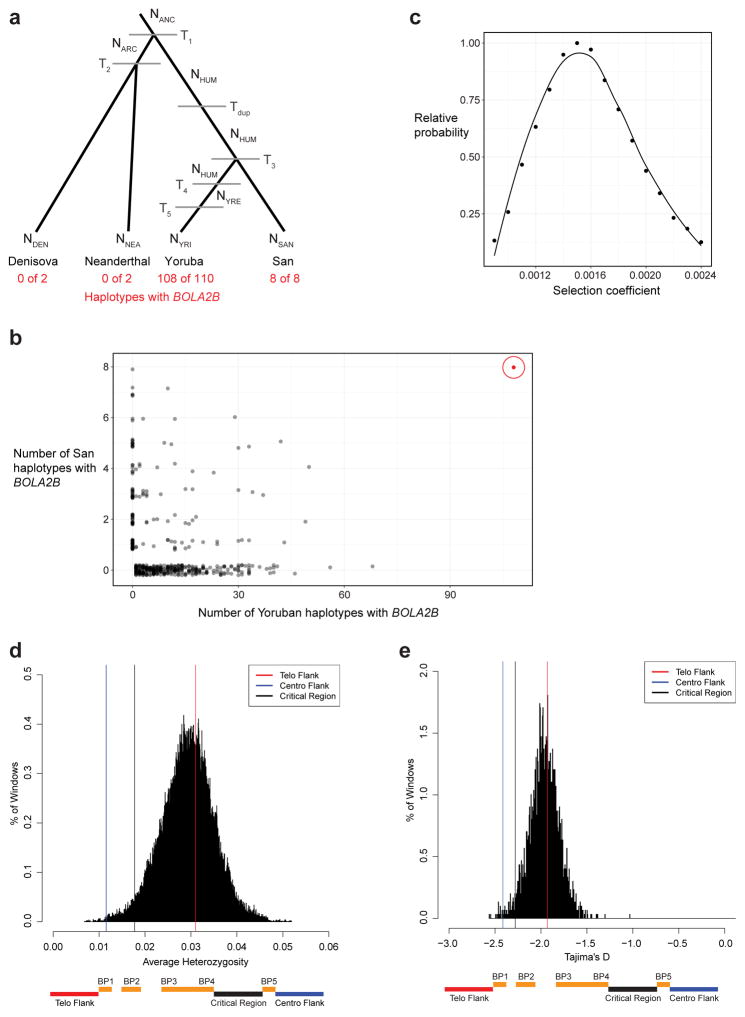
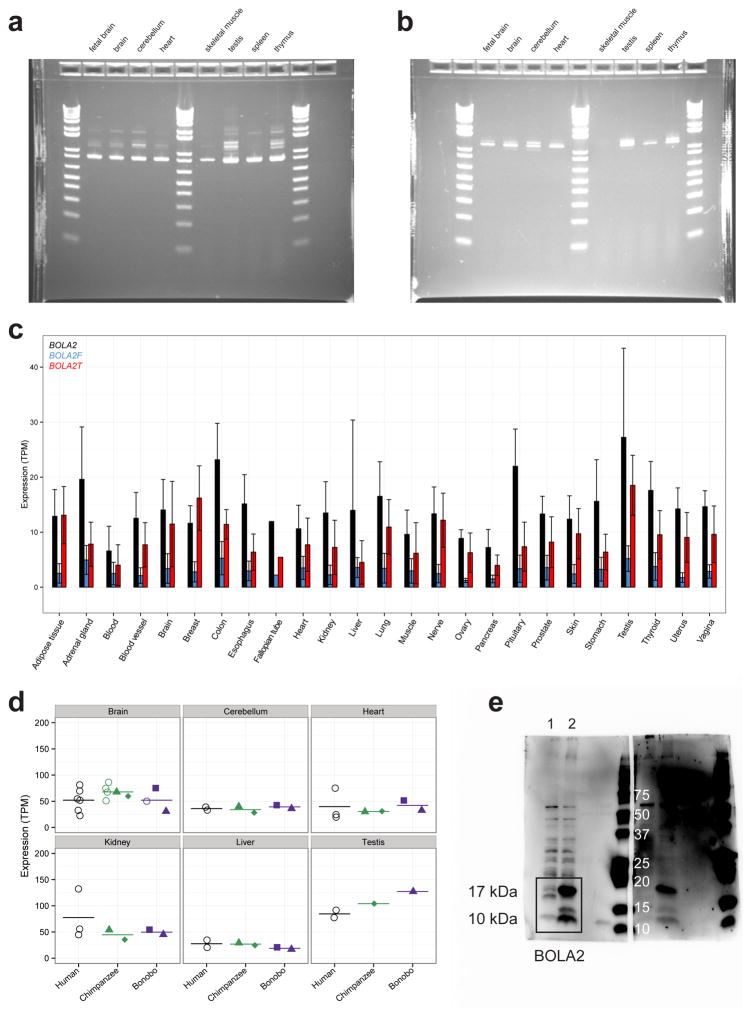
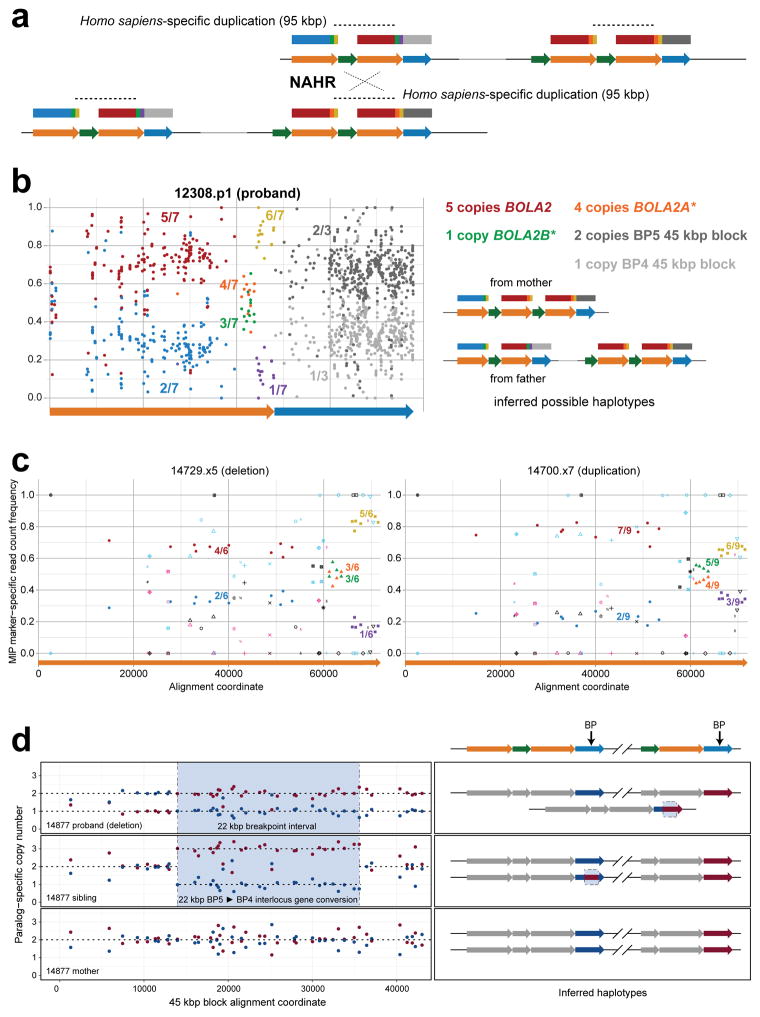
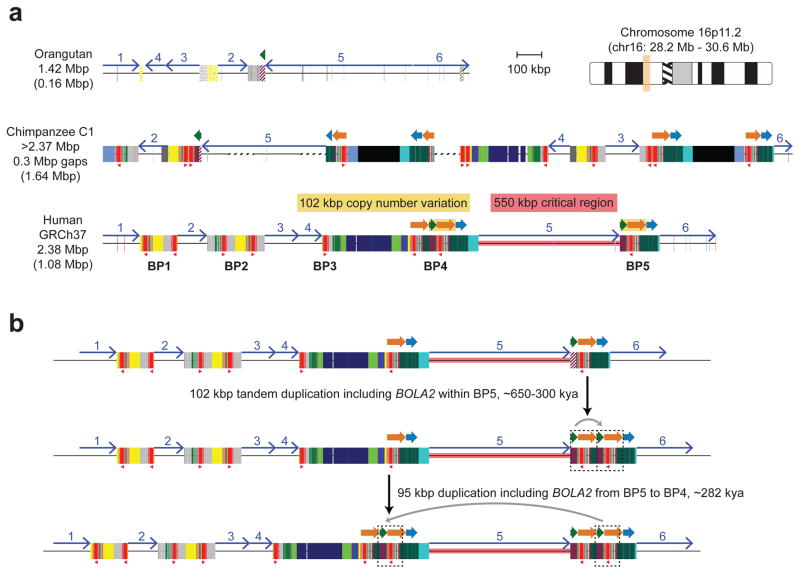
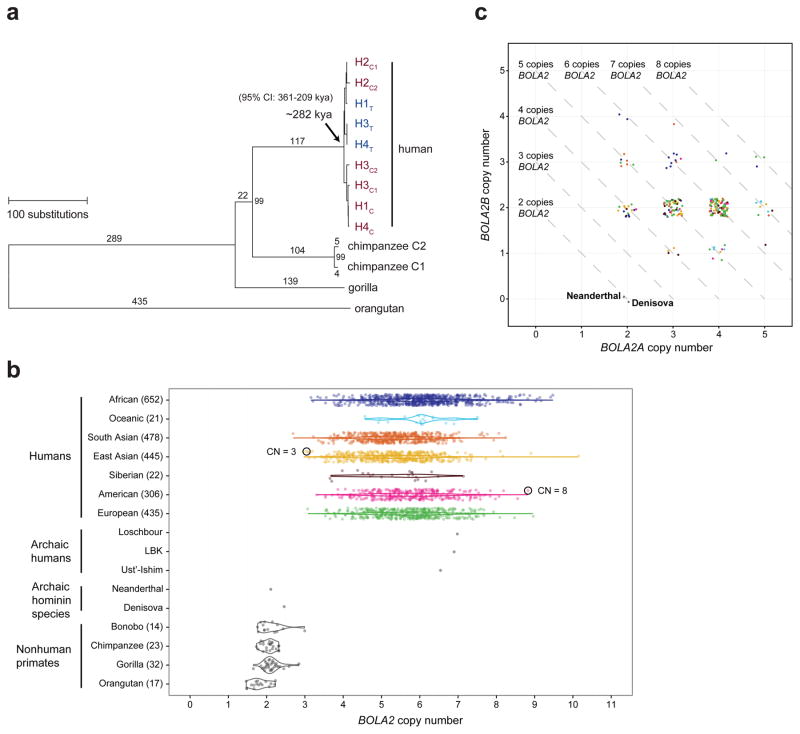
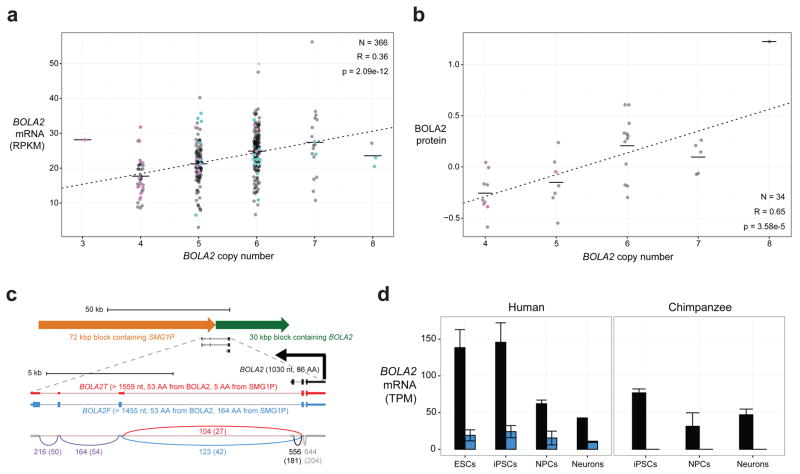
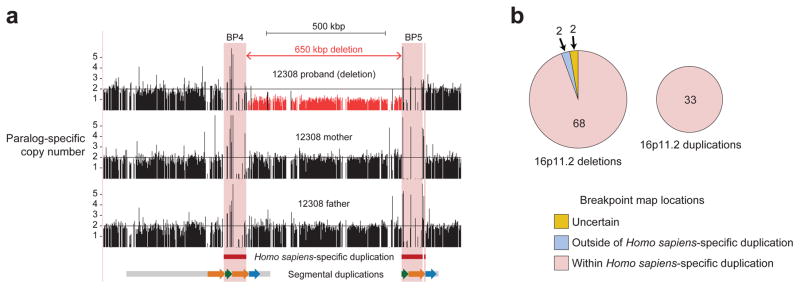
Genetic differences that specify unique aspects of human evolution have typically been identified by comparative analyses between the genomes of humans and closely related primates, including more recently the genomes of archaic hominins. Not all regions of the genome, however, are equally amenable to such study. Recurrent copy number variation (CNV) at chromosome 16p11.2 accounts for approximately 1% of cases of autism and is mediated by a complex set of segmental duplications, many of which arose recently during human evolution. Here we reconstruct the evolutionary history of the locus and identify bolA family member 2 (BOLA2) as a gene duplicated exclusively in Homo sapiens. We estimate that a 95-kilobase-pair segment containing BOLA2 duplicated across the critical region approximately 282 thousand years ago (ka), one of the latest among a series of genomic changes that dramatically restructured the locus during hominid evolution. All humans examined carried one or more copies of the duplication, which nearly fixed early in the human lineage--a pattern unlikely to have arisen so rapidly in the absence of selection (P < 0.0097). We show that the duplication of BOLA2 led to a novel, human-specific in-frame fusion transcript and that BOLA2 copy number correlates with both RNA expression (r = 0.36) and protein level (r = 0.65), with the greatest expression difference between human and chimpanzee in experimentally derived stem cells. Analyses of 152 patients carrying a chromosome 16p11. rearrangement show that more than 96% of breakpoints occur within the H. sapiens-specific duplication. In summary, the duplicative transposition of BOLA2 at the root of the H. sapiens lineage about 282 ka simultaneously increased copy number of a gene associated with iron homeostasis and predisposed our species to recurrent rearrangements associated with disease.
Conflict of interest statement
E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc., and is a consultant for the Kunming University of Science and Technology (KUST) as part of the 1000 China Talent Program.
Figures
References
-
- King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science (New York, NY) 1975;188:107–116. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
