

ABSSeq: a new RNA-Seq analysis method based on modelling absolute expression differences
- PMID: 27488180
- PMCID: PMC4973090
- DOI: 10.1186/s12864-016-2848-2
ABSSeq: a new RNA-Seq analysis method based on modelling absolute expression differences
Abstract
Background: The recent advances in next generation sequencing technology have made the sequencing of RNA (i.e., RNA-Seq) an extemely popular approach for gene expression analysis. Identification of significant differential expression represents a crucial initial step in these analyses, on which most subsequent inferences of biological functions are built. Yet, for identification of these subsequently analysed genes, most studies use an additional minimal threshold of differential expression that is not captured by the applied statistical procedures.
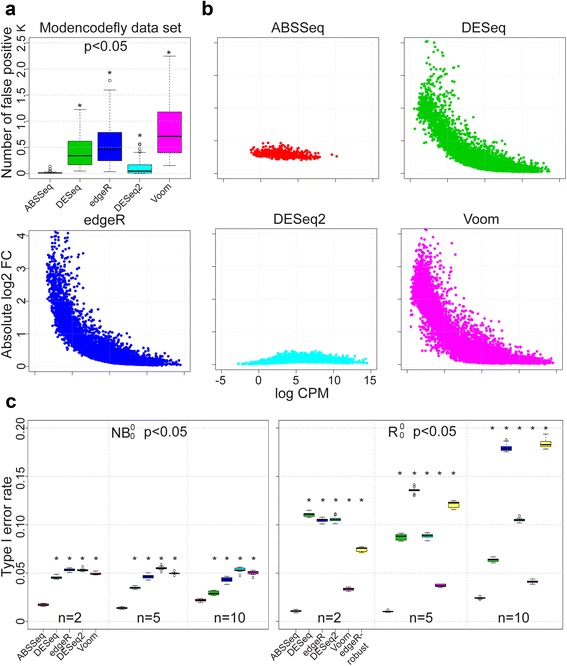
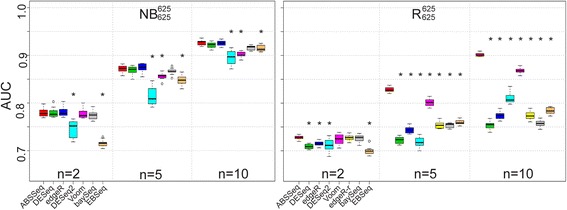
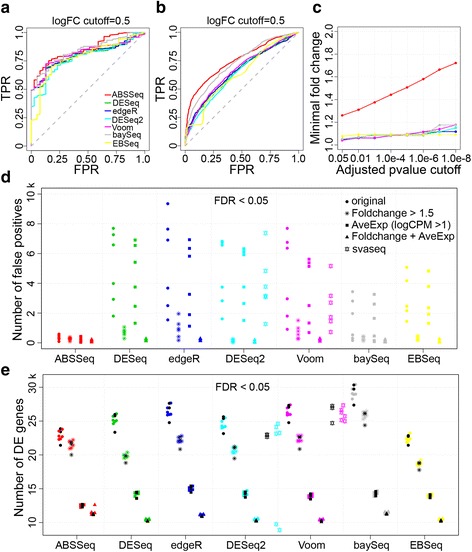
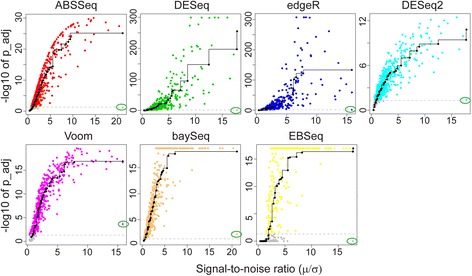
Results: Here we introduce a new analysis approach, ABSSeq, which uses a negative binomal distribution to model absolute expression differences between conditions, taking into account variations across genes and samples as well as magnitude of differences. In comparison to alternative methods, ABSSeq shows higher performance on controling type I error rate and at least a similar ability to correctly identify differentially expressed genes.
Conclusions: ABSSeq specifically considers the overall magnitude of expression differences, which enhances the power in detecting truly differentially expressed genes by reducing false positives at both very low and high expression level. In addition, ABSSeq offers to calculate shrinkage of fold change to facilitate gene ranking and effective outlier detection.
Keywords: ABSSeq; Differential gene expression; Negative binomial distribution; RNA-Seq; Transcriptome analysis.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
