

Combination Therapies for Lysosomal Storage Diseases: A Complex Answer to a Simple Problem
- PMID: 27491211
- PMCID: PMC5374980
Combination Therapies for Lysosomal Storage Diseases: A Complex Answer to a Simple Problem
Abstract
Abstract Lysosomal storage diseases (LSDs) are a group of 40-50 rare monogenic disorders that result in disrupted lysosomal function and subsequent lysosomal pathology. Depending on the protein or enzyme deficiency associated with each disease, LSDs affect an array of organ systems and elicit a complex set of secondary disease mechanisms that make many of these disorders difficult to fully treat. The etiology of most LSDs is known and the innate biology of lysosomal enzymes favors therapeutic intervention, yet most attempts at treating LSDs with enzyme replacement strategies fall short of being curative. Even with the advent of more sophisticated approaches, like substrate reduction therapy, pharmacologic chaperones, gene therapy or stem cell therapy, comprehensive treatments for LSDs have yet to be achieved. Given the limitations with individual therapies, recent research has focused on using a combination approach to treat LSDs. By coupling protein-, cell-, and gene- based therapies with small molecule drugs, researchers have found greater success in eradicating the clinical features of disease. This review seeks to discuss the positive and negatives of singular therapies used to treat LSDs, and discuss how, in combination, studies have demonstrated a more holistic benefit on pathological and functional parameters. By optimizing routes of delivery, therapeutic timing, and targeting secondary disease mechanisms, combination therapy represents the future for LSD treatment.
Figures
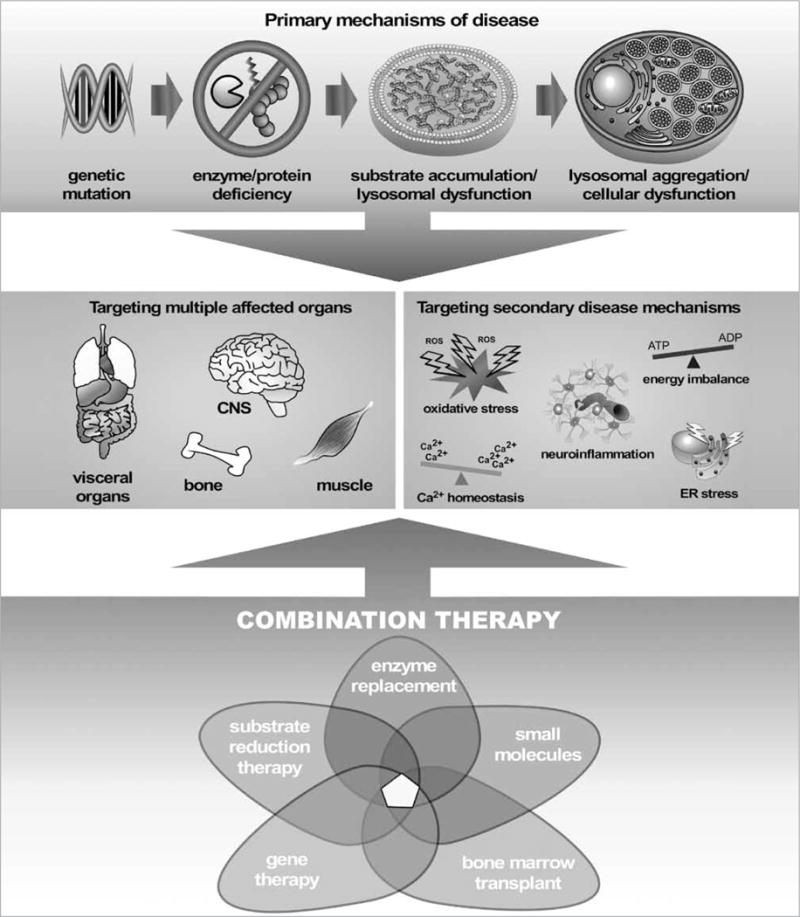
Similar articles
-
New strategies for the treatment of lysosomal storage diseases (review).Int J Mol Med. 2013 Jan;31(1):11-20. doi: 10.3892/ijmm.2012.1187. Epub 2012 Nov 19. Int J Mol Med. 2013. PMID: 23165354 Review.
-
Treatment of lysosomal storage diseases: recent patents and future strategies.Recent Pat Endocr Metab Immune Drug Discov. 2014 Jan;8(1):9-25. doi: 10.2174/1872214808666140115111350. Recent Pat Endocr Metab Immune Drug Discov. 2014. PMID: 24433521 Review.
-
Treatment options for lysosomal storage disorders: developing insights.Expert Opin Pharmacother. 2012 Nov;13(16):2281-99. doi: 10.1517/14656566.2012.729039. Epub 2012 Sep 26. Expert Opin Pharmacother. 2012. PMID: 23009070 Review.
-
Enzyme Replacement Therapy: A Review and Its Role in Treating Lysosomal Storage Diseases.Pediatr Ann. 2018 May 1;47(5):e191-e197. doi: 10.3928/19382359-20180424-01. Pediatr Ann. 2018. PMID: 29750286 Review.
-
Treatment strategies for lysosomal storage disorders.Dev Med Child Neurol. 2018 Jan;60(1):13-18. doi: 10.1111/dmcn.13600. Epub 2017 Nov 1. Dev Med Child Neurol. 2018. PMID: 29090451 Review.
Cited by
-
Elucidating the functional impact of G137V and G144R variants in Maroteaux Lamy's Syndrome by Molecular Dynamics Simulation.Mol Divers. 2024 Aug;28(4):2049-2063. doi: 10.1007/s11030-023-10694-8. Epub 2023 Jul 17. Mol Divers. 2024. PMID: 37458922
-
Precision Medicine for Lysosomal Disorders.Biomolecules. 2020 Jul 26;10(8):1110. doi: 10.3390/biom10081110. Biomolecules. 2020. PMID: 32722587 Free PMC article. Review.
-
Targeting the central nervous system in lysosomal storage diseases: Strategies to deliver therapeutics across the blood-brain barrier.Mol Ther. 2023 Mar 1;31(3):657-675. doi: 10.1016/j.ymthe.2022.11.015. Epub 2022 Nov 30. Mol Ther. 2023. PMID: 36457248 Free PMC article. Review.
-
C. elegans-based screen identifies lysosome-damaging alkaloids that induce STAT3-dependent lysosomal cell death.Protein Cell. 2018 Dec;9(12):1013-1026. doi: 10.1007/s13238-018-0520-0. Epub 2018 Apr 2. Protein Cell. 2018. PMID: 29611115 Free PMC article.
-
Mechanistic Insights into the Chaperoning of Human Lysosomal-Galactosidase Activity: Highly Functionalized Aminocyclopentanes and C-5a-Substituted Derivatives of 4-epi-Isofagomine.Molecules. 2020 Sep 3;25(17):4025. doi: 10.3390/molecules25174025. Molecules. 2020. PMID: 32899288 Free PMC article.
References
-
- Hers HG. Inborn Lysosomal Diseases. Gastroenterology. 1965;48:625–633. - PubMed
-
- Neufeld EF. Lysosomal storage diseases. Annu Rev Biochem. 1991;60:257–280. - PubMed
-
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders JAMA. 1999;281:249–254. - PubMed
-
- Scriver CR. The Metabolic and Molecular Basis of Inherited Disease. New York: McGraw-Hill; 2001. p. 7012.
-
- Winchester B. Primary defects in lysosomal enzymes. Oxford: Oxford University Press; 2004. p. 81.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical