

Recognizing millions of consistently unidentified spectra across hundreds of shotgun proteomics datasets
- PMID: 27493588
- PMCID: PMC4968634
- DOI: 10.1038/nmeth.3902
Recognizing millions of consistently unidentified spectra across hundreds of shotgun proteomics datasets
Abstract
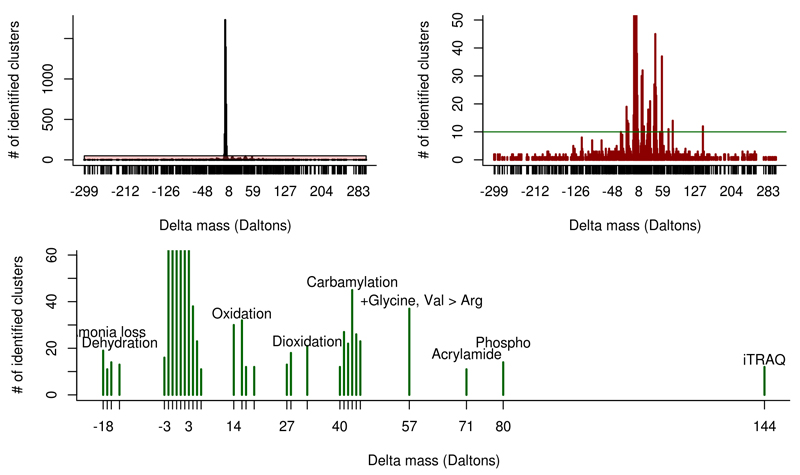
Mass spectrometry (MS) is the main technology used in proteomics approaches. However, on average 75% of spectra analysed in an MS experiment remain unidentified. We propose to use spectrum clustering at a large-scale to shed a light on these unidentified spectra. PRoteomics IDEntifications database (PRIDE) Archive is one of the largest MS proteomics public data repositories worldwide. By clustering all tandem MS spectra publicly available in PRIDE Archive, coming from hundreds of datasets, we were able to consistently characterize three distinct groups of spectra: 1) incorrectly identified spectra, 2) spectra correctly identified but below the set scoring threshold, and 3) truly unidentified spectra. Using a multitude of complementary analysis approaches, we were able to identify less than 20% of the consistently unidentified spectra. The complete spectrum clustering results are available through the new version of the PRIDE Cluster resource (http://www.ebi.ac.uk/pride/cluster). This resource is intended, among other aims, to encourage and simplify further investigation into these unidentified spectra.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
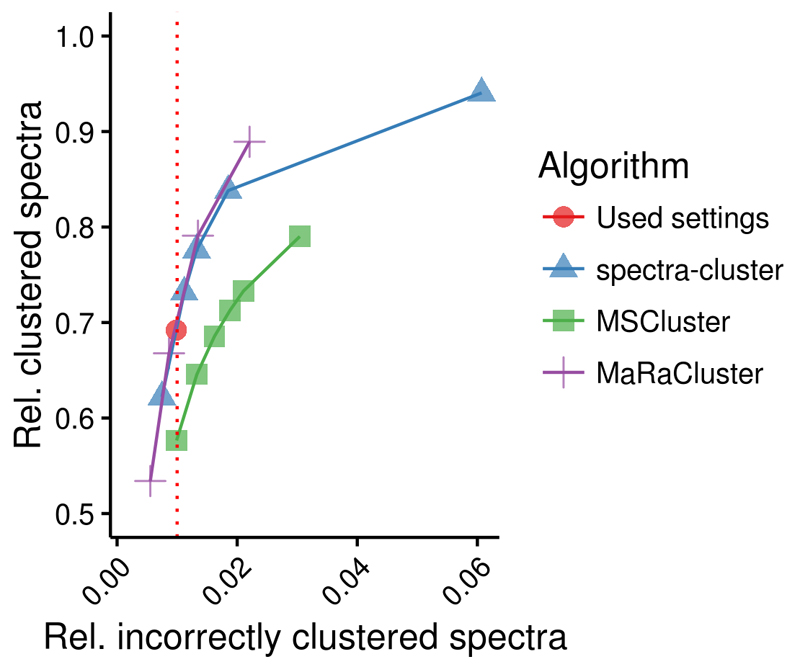
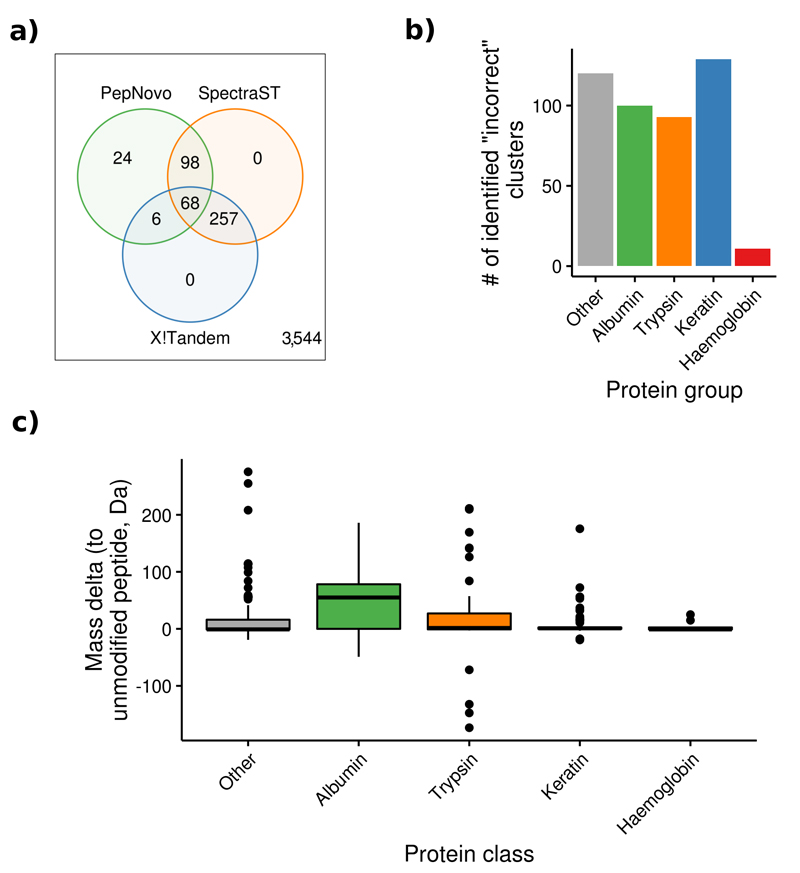
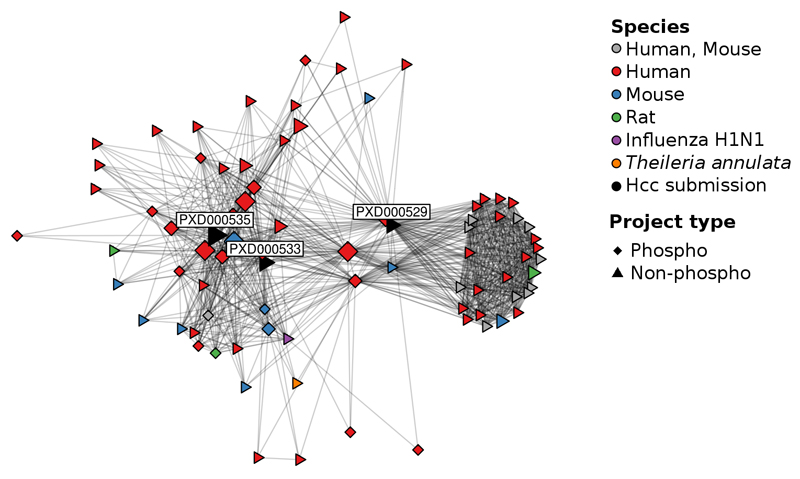
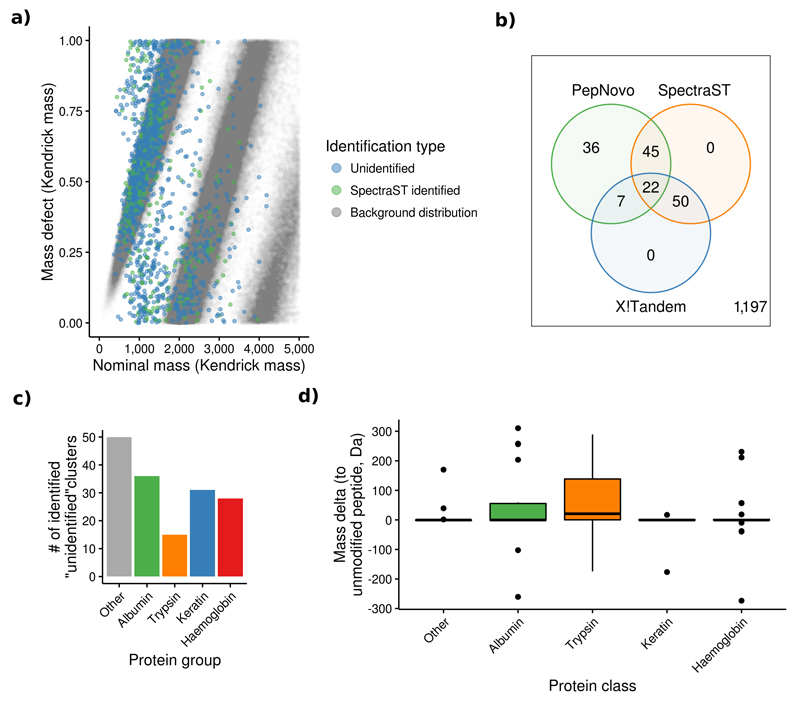
References
-
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. - PubMed
-
- Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994;5:976–989. - PubMed
-
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. - PubMed
-
- Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
