

Modeling protein-DNA binding via high-throughput in vitro technologies
- PMID: 27497616
- PMCID: PMC5439287
- DOI: 10.1093/bfgp/elw030
Modeling protein-DNA binding via high-throughput in vitro technologies
Abstract
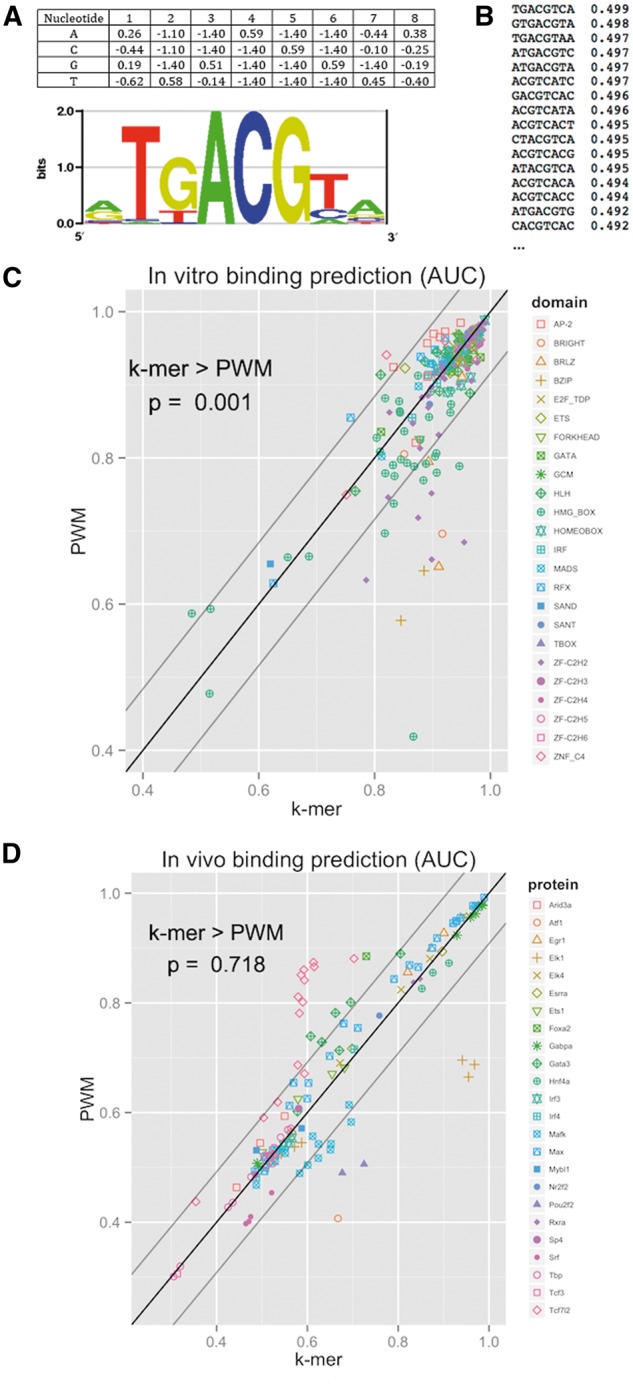
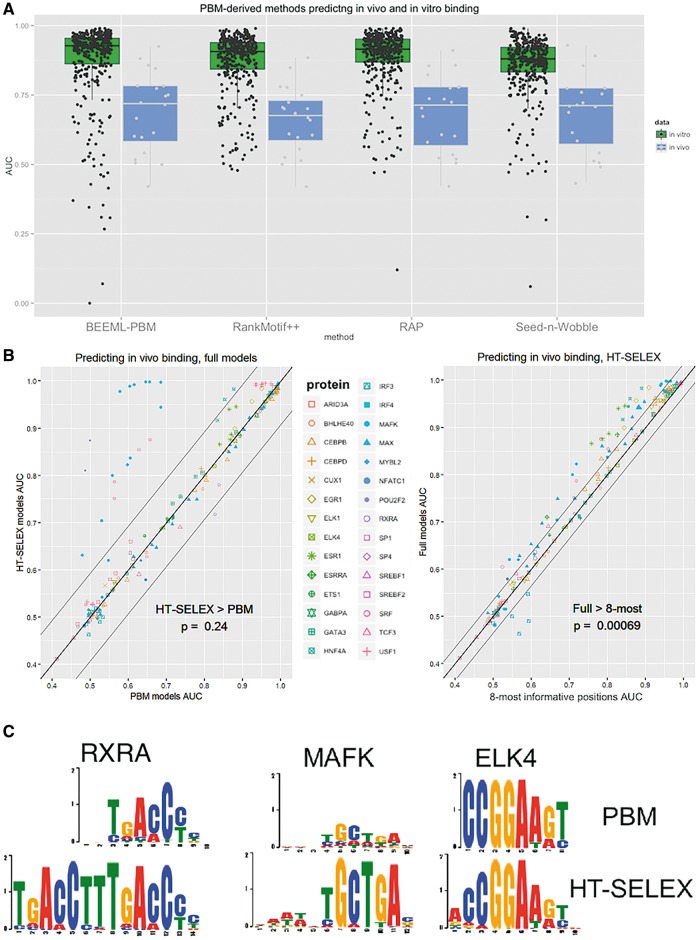
Protein-DNA binding plays a central role in gene regulation and by that in all processes in the living cell. Novel experimental and computational approaches facilitate better understanding of protein-DNA binding preferences via high-throughput measurement of protein binding to a large number of DNA sequences and inference of binding models from them. Here we review the state of the art in measuring protein-DNA binding in vitro, emphasizing the advantages and limitations of different technologies. In addition, we describe models for representing protein-DNA binding preferences and key computational approaches to learn those from high-throughput data. Using large experimental data sets, we test the performance of different models based on different measuring techniques. We conclude with pertinent open problems.
Keywords: high-throughput SELEX; motif finding; protein-binding microarrays; protein–DNA binding.
© The Author 2016. Published by Oxford University Press.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
