

The Power of Human Protective Modifiers: PLS3 and CORO1C Unravel Impaired Endocytosis in Spinal Muscular Atrophy and Rescue SMA Phenotype
- PMID: 27499521
- PMCID: PMC5011078
- DOI: 10.1016/j.ajhg.2016.07.014
The Power of Human Protective Modifiers: PLS3 and CORO1C Unravel Impaired Endocytosis in Spinal Muscular Atrophy and Rescue SMA Phenotype
Abstract
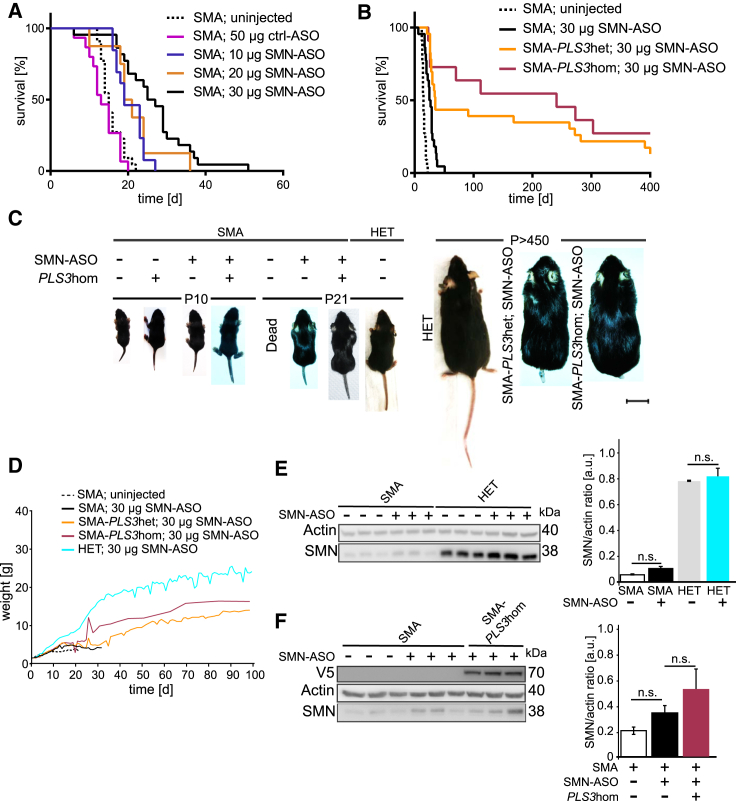
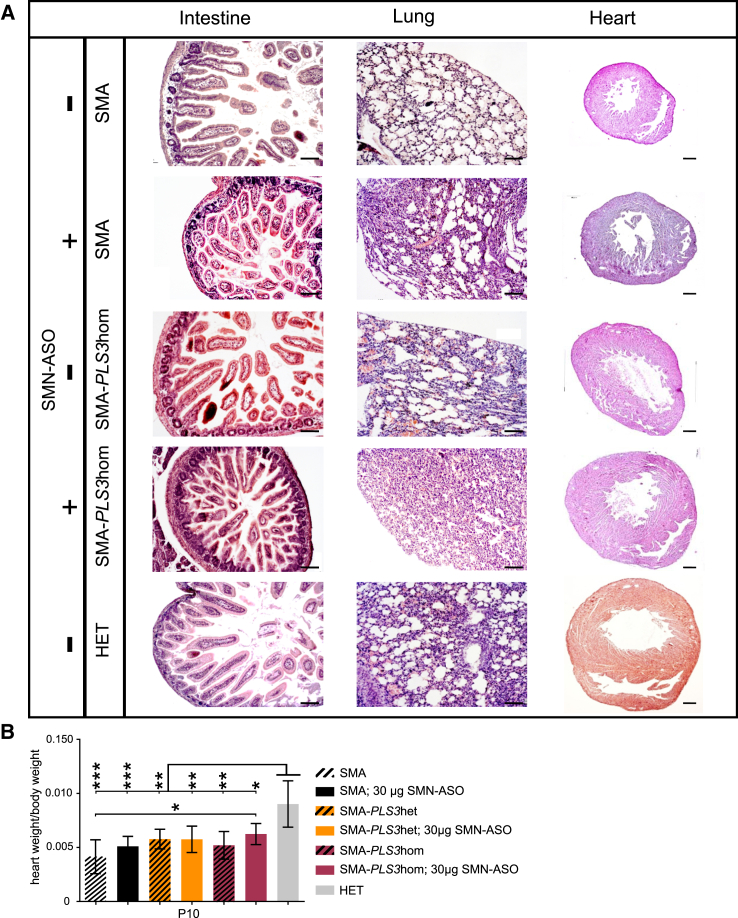
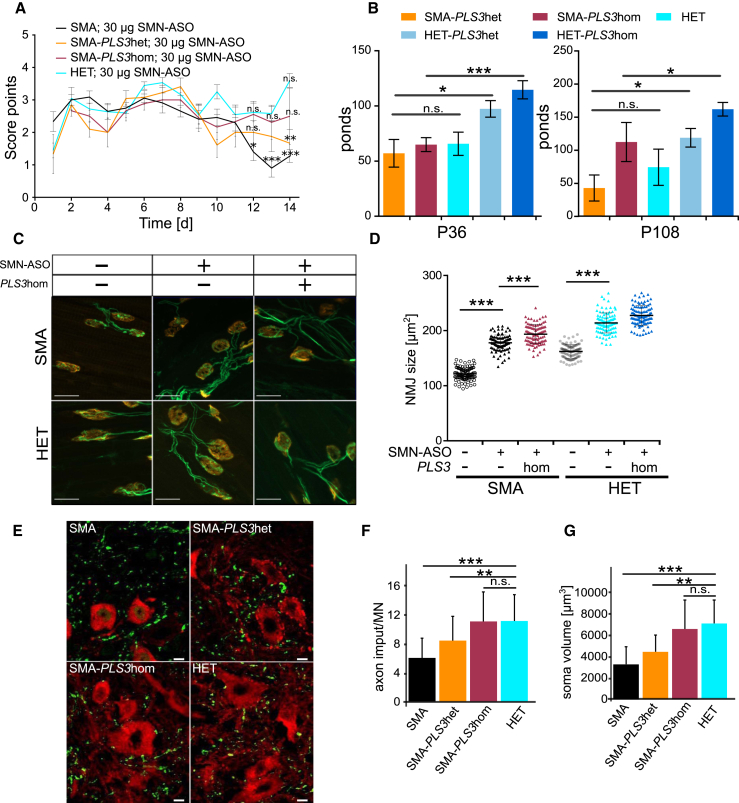
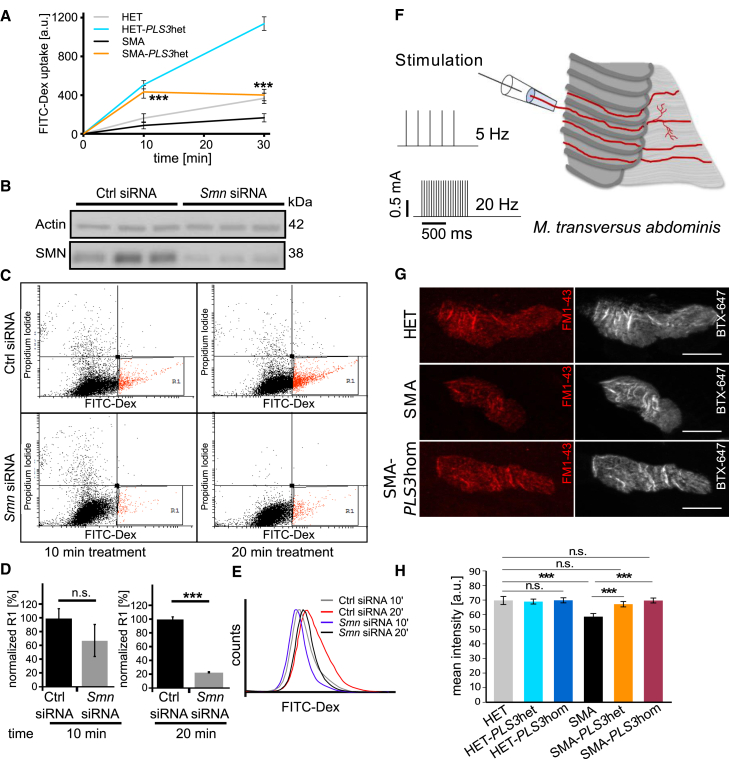
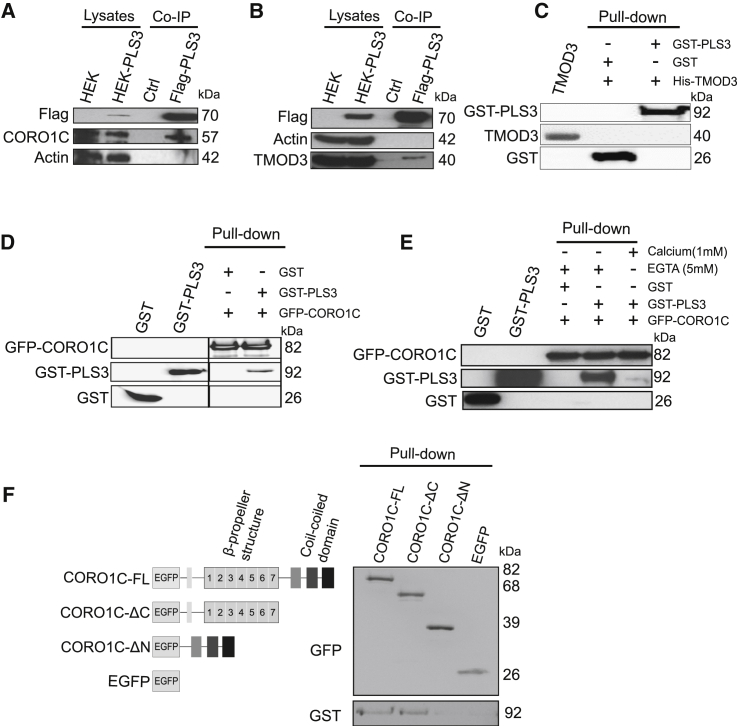
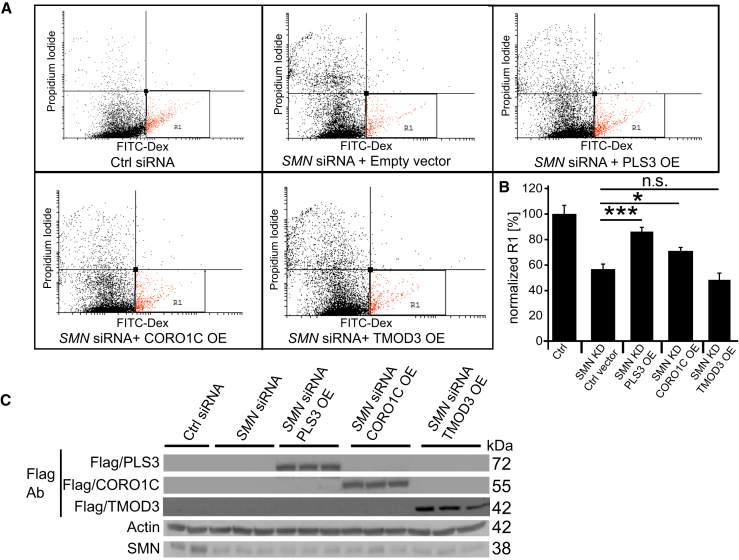
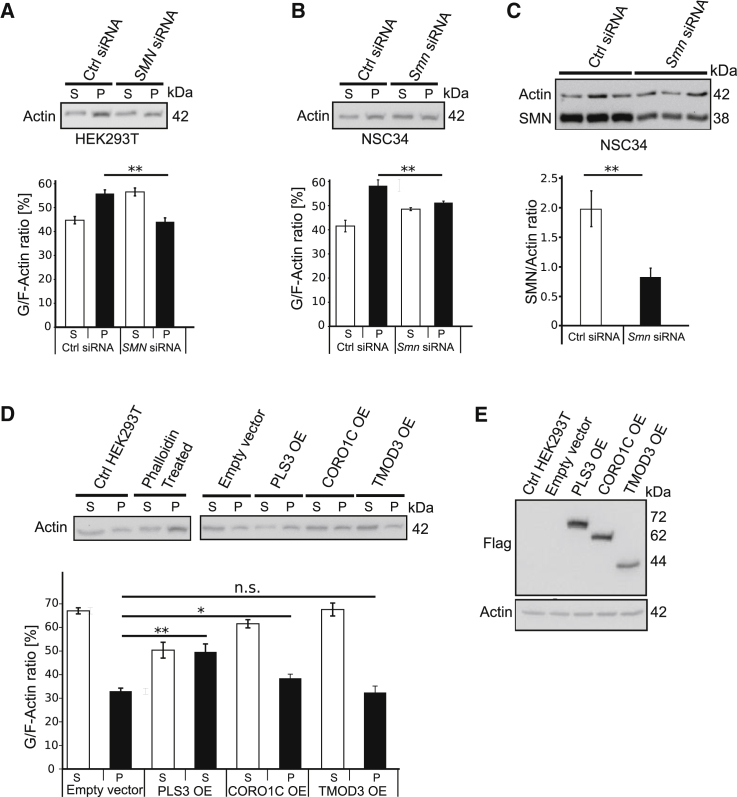
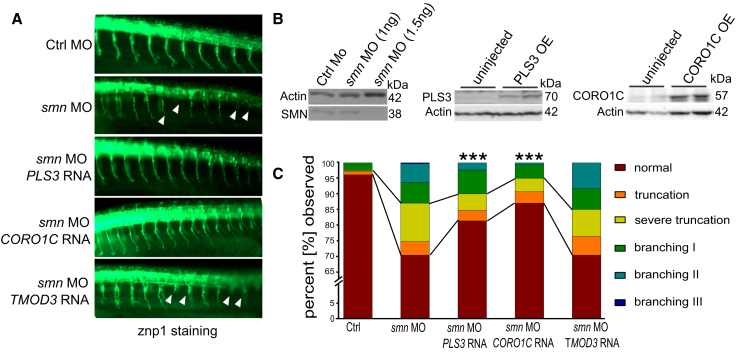
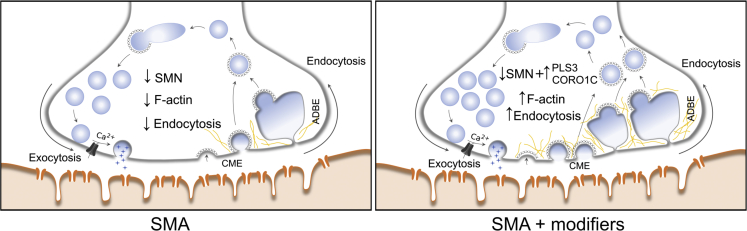
Homozygous loss of SMN1 causes spinal muscular atrophy (SMA), the most common and devastating childhood genetic motor-neuron disease. The copy gene SMN2 produces only ∼10% functional SMN protein, insufficient to counteract development of SMA. In contrast, the human genetic modifier plastin 3 (PLS3), an actin-binding and -bundling protein, fully protects against SMA in SMN1-deleted individuals carrying 3-4 SMN2 copies. Here, we demonstrate that the combinatorial effect of suboptimal SMN antisense oligonucleotide treatment and PLS3 overexpression-a situation resembling the human condition in asymptomatic SMN1-deleted individuals-rescues survival (from 14 to >250 days) and motoric abilities in a severe SMA mouse model. Because PLS3 knockout in yeast impairs endocytosis, we hypothesized that disturbed endocytosis might be a key cellular mechanism underlying impaired neurotransmission and neuromuscular junction maintenance in SMA. Indeed, SMN deficit dramatically reduced endocytosis, which was restored to normal levels by PLS3 overexpression. Upon low-frequency electro-stimulation, endocytotic FM1-43 (SynaptoGreen) uptake in the presynaptic terminal of neuromuscular junctions was restored to control levels in SMA-PLS3 mice. Moreover, proteomics and biochemical analysis revealed CORO1C, another F-actin binding protein, whose direct binding to PLS3 is dependent on calcium. Similar to PLS3 overexpression, CORO1C overexpression restored fluid-phase endocytosis in SMN-knockdown cells by elevating F-actin amounts and rescued the axonal truncation and branching phenotype in Smn-depleted zebrafish. Our findings emphasize the power of genetic modifiers to unravel the cellular pathomechanisms underlying SMA and the power of combinatorial therapy based on splice correction of SMN2 and endocytosis improvement to efficiently treat SMA.
Copyright © 2016 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
CHP1 reduction ameliorates spinal muscular atrophy pathology by restoring calcineurin activity and endocytosis.Brain. 2018 Aug 1;141(8):2343-2361. doi: 10.1093/brain/awy167. Brain. 2018. PMID: 29961886 Free PMC article.
-
Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis.Am J Hum Genet. 2017 Feb 2;100(2):297-315. doi: 10.1016/j.ajhg.2017.01.005. Epub 2017 Jan 26. Am J Hum Genet. 2017. PMID: 28132687 Free PMC article.
-
Calcium binding is essential for plastin 3 function in Smn-deficient motoneurons.Hum Mol Genet. 2014 Apr 15;23(8):1990-2004. doi: 10.1093/hmg/ddt595. Epub 2013 Nov 23. Hum Mol Genet. 2014. PMID: 24271012 Free PMC article.
-
New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand?Int J Mol Sci. 2020 May 7;21(9):3297. doi: 10.3390/ijms21093297. Int J Mol Sci. 2020. PMID: 32392694 Free PMC article. Review.
-
Plastin 3 in health and disease: a matter of balance.Cell Mol Life Sci. 2021 Jul;78(13):5275-5301. doi: 10.1007/s00018-021-03843-5. Epub 2021 May 23. Cell Mol Life Sci. 2021. PMID: 34023917 Free PMC article. Review.
Cited by
-
Calcium is reduced in presynaptic mitochondria of motor nerve terminals during neurotransmission in SMA mice.Hum Mol Genet. 2021 May 17;30(8):629-643. doi: 10.1093/hmg/ddab065. Hum Mol Genet. 2021. PMID: 33693569 Free PMC article.
-
Biomarkers and the Development of a Personalized Medicine Approach in Spinal Muscular Atrophy.Front Neurol. 2019 Aug 19;10:898. doi: 10.3389/fneur.2019.00898. eCollection 2019. Front Neurol. 2019. PMID: 31481927 Free PMC article. Review.
-
NMJ-related diseases beyond the congenital myasthenic syndromes.Front Cell Dev Biol. 2023 Aug 4;11:1216726. doi: 10.3389/fcell.2023.1216726. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 37601107 Free PMC article. Review.
-
Systematic review and meta-analysis determining the benefits of in vivo genetic therapy in spinal muscular atrophy rodent models.Gene Ther. 2022 Sep;29(9):498-512. doi: 10.1038/s41434-021-00292-4. Epub 2021 Oct 6. Gene Ther. 2022. PMID: 34611322 Free PMC article.
-
Survival Motor Neuron Protein is Released from Cells in Exosomes: A Potential Biomarker for Spinal Muscular Atrophy.Sci Rep. 2017 Oct 24;7(1):13859. doi: 10.1038/s41598-017-14313-z. Sci Rep. 2017. PMID: 29066780 Free PMC article.
References
-
- Pellizzoni L., Kataoka N., Charroux B., Dreyfuss G. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell. 1998;95:615–624. - PubMed
-
- Liu Q., Fischer U., Wang F., Dreyfuss G. The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in a complex with spliceosomal snRNP proteins. Cell. 1997;90:1013–1021. - PubMed
-
- Lefebvre S., Bürglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
