

Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation
- PMID: 27500631
- PMCID: PMC5157049
- DOI: 10.1038/ni.3519
Plasma cell differentiation is coupled to division-dependent DNA hypomethylation and gene regulation
Abstract
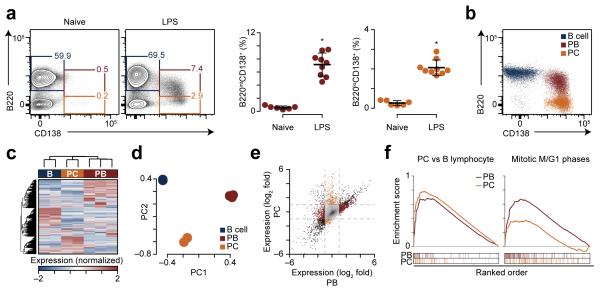
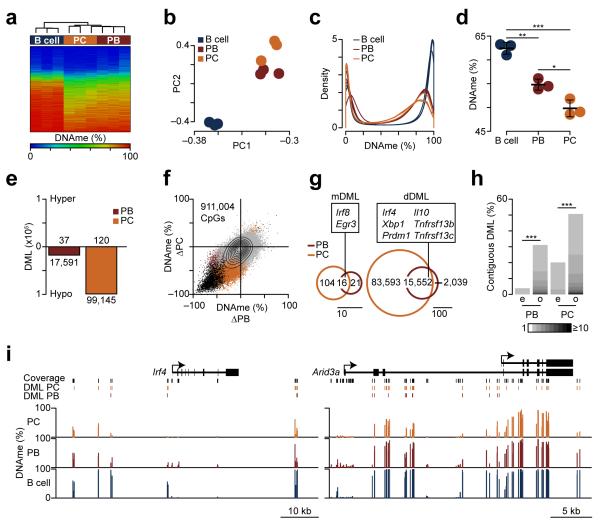
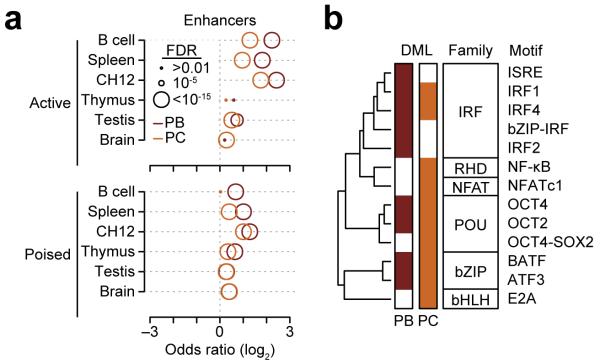
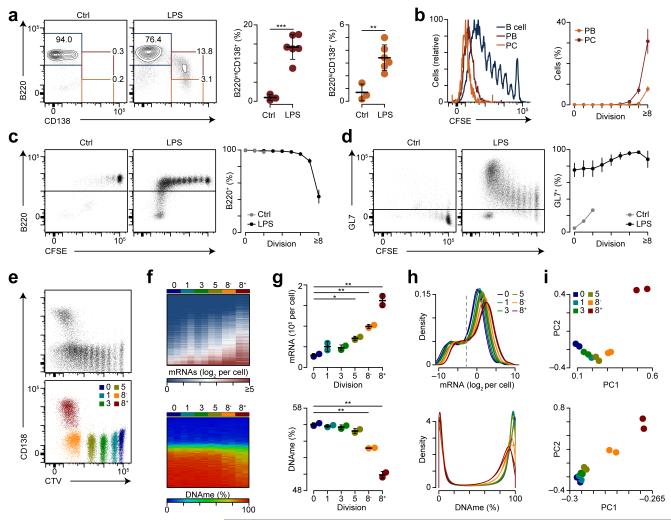
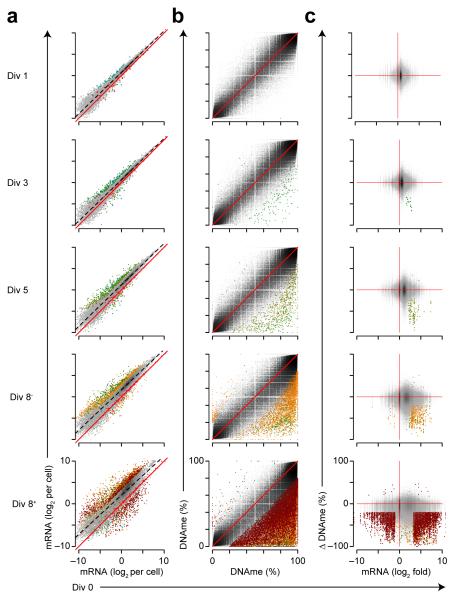
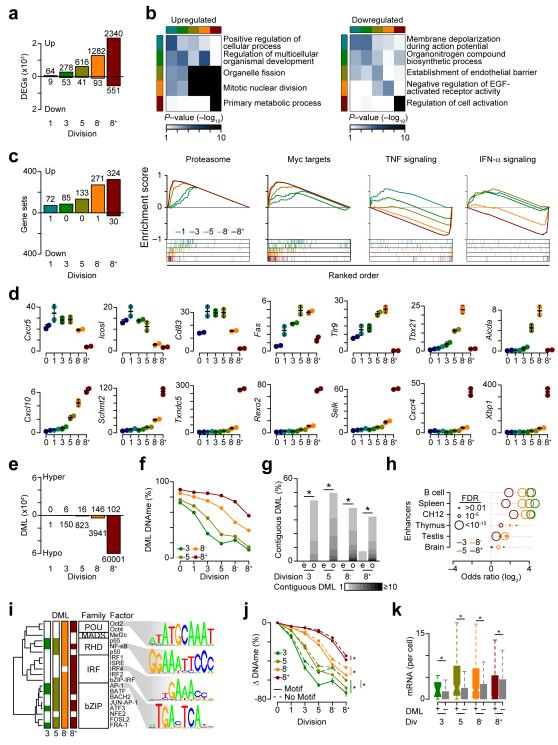
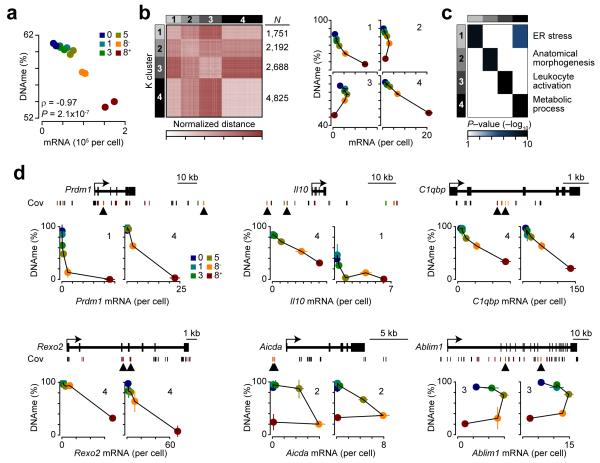
The epigenetic processes that regulate antibody-secreting plasma cells are not well understood. Here, analysis of plasma cell differentiation revealed DNA hypomethylation of 10% of CpG loci that were overrepresented at enhancers. Inhibition of DNA methylation enhanced plasma cell commitment in a cell-division-dependent manner. Analysis of B cells differentiating in vivo stratified by cell division revealed a fivefold increase in mRNA transcription coupled to DNA hypomethylation. Demethylation occurred first at binding motifs for the transcription factors NF-κB and AP-1 and later at those for the transcription factors IRF and Oct-2 and was coincident with activation and differentiation gene-expression programs in a cell-division-dependent manner. These data provide mechanistic insight into cell-division-coupled transcriptional and epigenetic reprogramming and suggest that DNA hypomethylation reflects the cis-regulatory history of plasma cell differentiation.
Figures

References
-
- Macallan DC, et al. B-cell kinetics in humans: rapid turnover of peripheral blood memory cells. Blood. 2005;105:3633–3640. - PubMed
-
- Nutt SL, Taubenheim N, Hasbold J, Corcoran LM, Hodgkin PD. The genetic network controlling plasma cell differentiation. 2011. - PubMed
-
- Hasbold J, Corcoran LM, Tarlinton DM, Tangye SG, Hodgkin PD. Evidence from the generation of immunoglobulin G–secreting cells that stochastic mechanisms regulate lymphocyte differentiation. Nat Immunol. 2004;5:55–63. - PubMed
Methods references
-
- Hsu F, et al. The UCSC Known Genes. Bioinformatics. 2006;22:1036–1046. - PubMed
-
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995;57:289–300.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
