

Prion-like propagation as a pathogenic principle in frontotemporal dementia
- PMID: 27502124
- PMCID: PMC6680357
- DOI: 10.1111/jnc.13668
Prion-like propagation as a pathogenic principle in frontotemporal dementia
Abstract
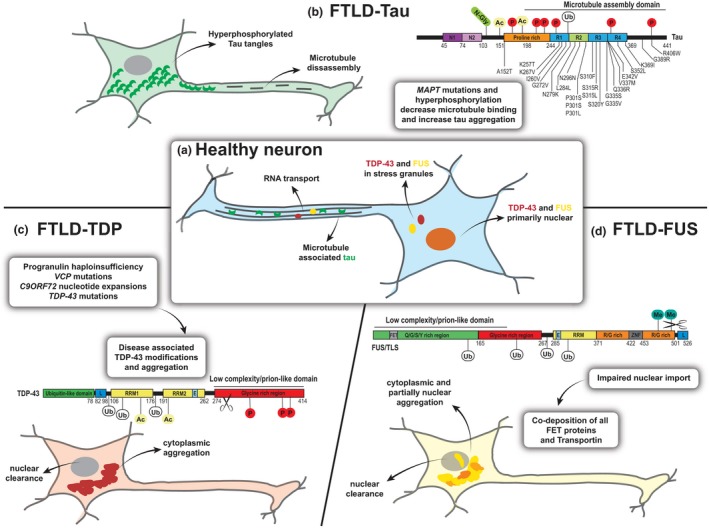
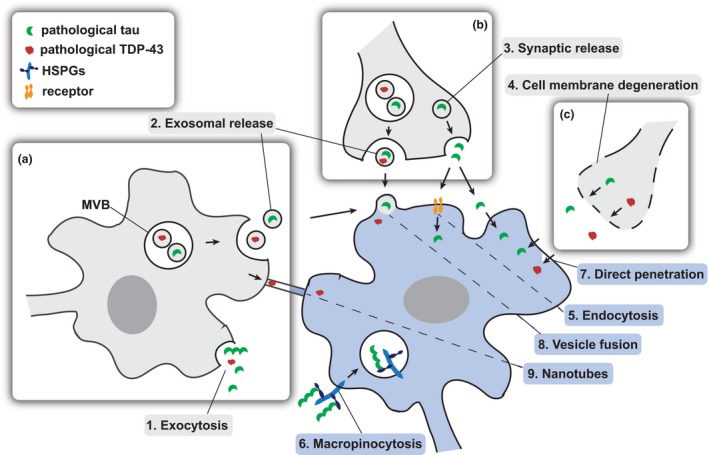
Frontotemporal dementia is a devastating neurodegenerative disease causing stark alterations in personality and language. Characterized by severe atrophy of the frontal and temporal brain lobes, frontotemporal dementia (FTD) shows extreme heterogeneity in clinical presentation, genetic causes, and pathological findings. Like most neurodegenerative diseases, the initial symptoms of FTD are subtle, but increase in severity over time, as the disease progresses. Clinical progression is paralleled by exacerbation of pathological findings and the involvement of broader brain regions, which currently lack mechanistic explanation. Yet, a flurry of studies indicate that protein aggregates accumulating in neurodegenerative diseases can act as propagating entities, amplifying their pathogenic conformation, in a way similar to infectious prions. In this prion-centric view, FTD can be divided into three subtypes, TDP-43 or FUS proteinopathy and tauopathy. Here, we review the current evidence that FTD-linked pathology propagates in a prion-like manner and discuss the implications of these findings for disease progression and heterogeneity. Frontotemporal dementia (FTD) is a progressive neurodegenerative disease causing severe personality dysfunctions, characterized by profound heterogeneity. Accumulation of tau, TDP-43 or FUS cytoplasmic aggregates characterize molecularly distinct and non-overlapping FTD subtypes. Here, we discuss the current evidence suggesting that prion-like propagation and cell-to-cell spread of each of these cytoplasmic aggregates may underlie disease progression and heterogeneity. This article is part of the Frontotemporal Dementia special issue.
Keywords: FTD; FUS; TDP-43; prion-like spread; tau.
© 2016 The Authors. Journal of Neurochemistry published by John Wiley & Sons Ltd on behalf of International Society for Neurochemistry.
Figures
References
-
- Agosta F., Scola E., Canu E. et al (2012) White matter damage in frontotemporal lobar degeneration spectrum. Cereb. Cortex 22, 2705–2714. - PubMed
-
- Aguzzi A. (2009) Cell biology: beyond the prion principle. Nature 459, 924–925. - PubMed
-
- Aguzzi A. and Polymenidou M. (2004) Mammalian prion biology: one century of evolving concepts. Cell 116, 313–327. - PubMed
-
- Aguzzi A. and Rajendran L. (2009) The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 64, 783–790. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
