

The Current Landscape of Genetic Testing in Cardiovascular Malformations: Opportunities and Challenges
- PMID: 27504451
- PMCID: PMC4959014
- DOI: 10.3389/fcvm.2016.00022
The Current Landscape of Genetic Testing in Cardiovascular Malformations: Opportunities and Challenges
Abstract
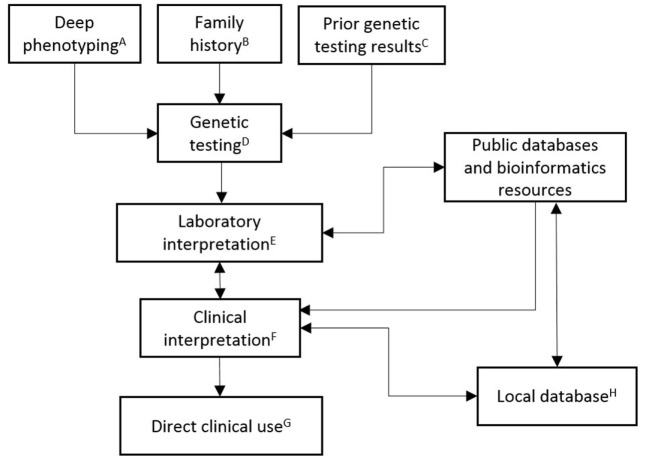
Human cardiovascular malformations (CVMs) frequently have a genetic contribution. Through the application of novel technologies, such as next-generation sequencing, DNA sequence variants associated with CVMs are being identified at a rapid pace. While clinicians are now able to offer testing with NGS gene panels or whole exome sequencing to any patient with a CVM, the interpretation of genetic variation remains problematic. Variable phenotypic expression, reduced penetrance, inconsistent phenotyping methods, and the lack of high-throughput functional testing of variants contribute to these challenges. This article elaborates critical issues that impact the decision to broadly implement clinical molecular genetic testing in CVMs. Major benefits of testing include establishing a genetic diagnosis, facilitating cost-effective screening of family members who may have subclinical disease, predicting recurrence risk in offsprings, enabling early diagnosis and anticipatory management of CV and non-CV disease phenotypes, predicting long-term outcomes, and facilitating the development of novel therapies aimed at disease improvement or prevention. Limitations include financial cost, psychosocial cost, and ambiguity of interpretation of results. Multiplex families and patients with syndromic features are two groups where disease causation could potentially be firmly established. However, these account for the minority of the overall CVM population, and there is increasing recognition that genotypes previously associated with syndromes also exist in patients who lack non-CV findings. In all circumstances, ongoing dialog between cardiologists and clinical geneticists will be needed to accurately interpret genetic testing and improve these patients' health. This may be most effectively implemented by the creation and support of CV genetics services at centers committed to pursuing testing for patients.
Keywords: congenital heart disease; genetics; genomics; mutation; next-generation sequencing; phenomics; phenotyping.
Figures
References
-
- Centers for Disease Control and Prevention (CDC). Hospital stays, hospital charges, and in-hospital deaths among infants with selected birth defects – United States, 2003. MMWR Morb Mortal Wkly Rep (2007) 56(2):25–9. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources