

Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis
- PMID: 27506793
- PMCID: PMC5003261
- DOI: 10.1073/pnas.1603244113
Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis
Abstract
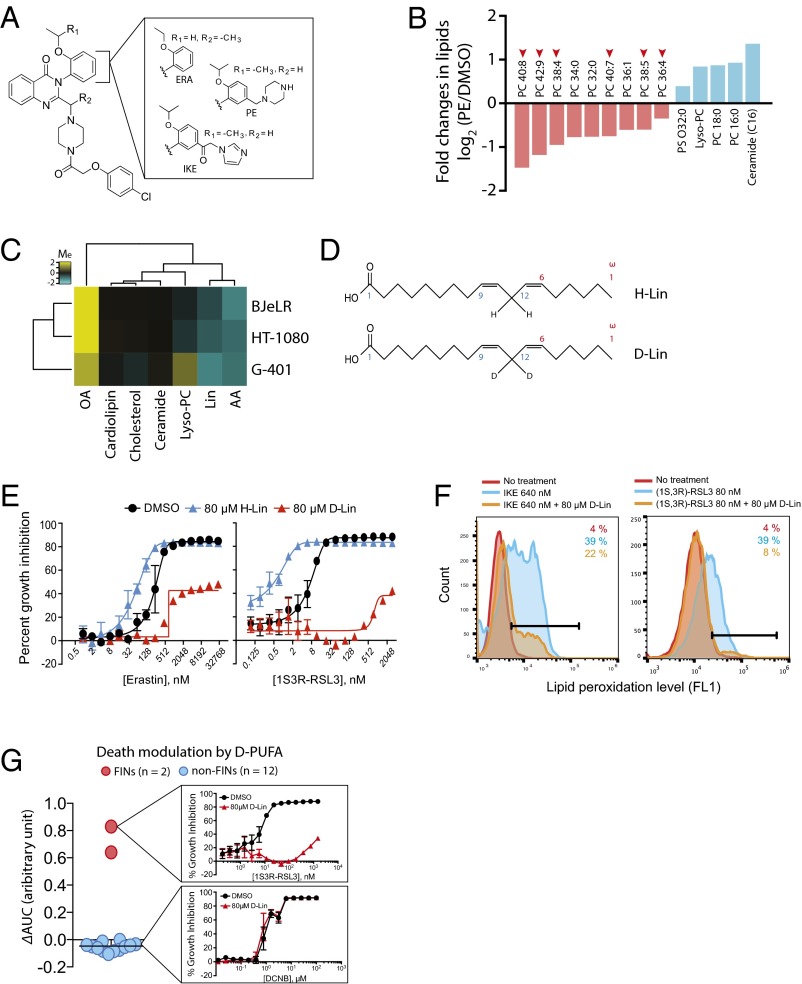
Ferroptosis is form of regulated nonapoptotic cell death that is involved in diverse disease contexts. Small molecules that inhibit glutathione peroxidase 4 (GPX4), a phospholipid peroxidase, cause lethal accumulation of lipid peroxides and induce ferroptotic cell death. Although ferroptosis has been suggested to involve accumulation of reactive oxygen species (ROS) in lipid environments, the mediators and substrates of ROS generation and the pharmacological mechanism of GPX4 inhibition that generates ROS in lipid environments are unknown. We report here the mechanism of lipid peroxidation during ferroptosis, which involves phosphorylase kinase G2 (PHKG2) regulation of iron availability to lipoxygenase enzymes, which in turn drive ferroptosis through peroxidation of polyunsaturated fatty acids (PUFAs) at the bis-allylic position; indeed, pretreating cells with PUFAs containing the heavy hydrogen isotope deuterium at the site of peroxidation (D-PUFA) prevented PUFA oxidation and blocked ferroptosis. We further found that ferroptosis inducers inhibit GPX4 by covalently targeting the active site selenocysteine, leading to accumulation of PUFA hydroperoxides. In summary, we found that PUFA oxidation by lipoxygenases via a PHKG2-dependent iron pool is necessary for ferroptosis and that the covalent inhibition of the catalytic selenocysteine in Gpx4 prevents elimination of PUFA hydroperoxides; these findings suggest new strategies for controlling ferroptosis in diverse contexts.
Keywords: Gpx4; PHKG2; PUFAs; ferroptosis; lipoxygenase.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






References
-
- Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285–296. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
