

Comparison of Fusarium graminearum Transcriptomes on Living or Dead Wheat Differentiates Substrate-Responsive and Defense-Responsive Genes
- PMID: 27507961
- PMCID: PMC4960244
- DOI: 10.3389/fmicb.2016.01113
Comparison of Fusarium graminearum Transcriptomes on Living or Dead Wheat Differentiates Substrate-Responsive and Defense-Responsive Genes
Abstract
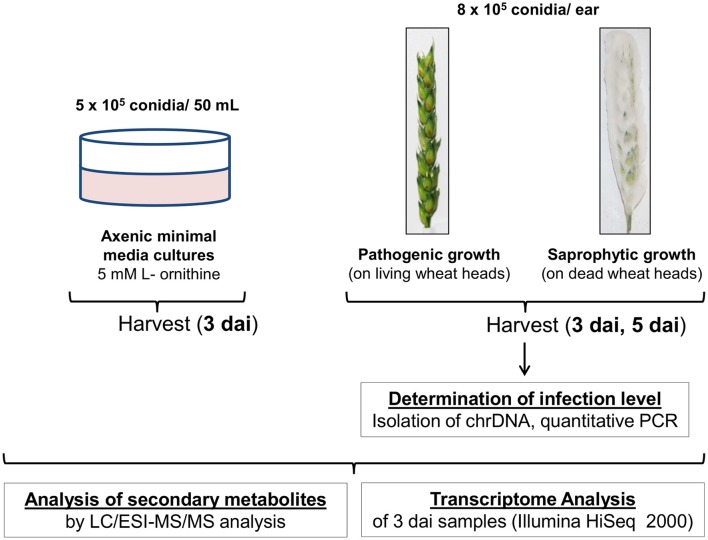
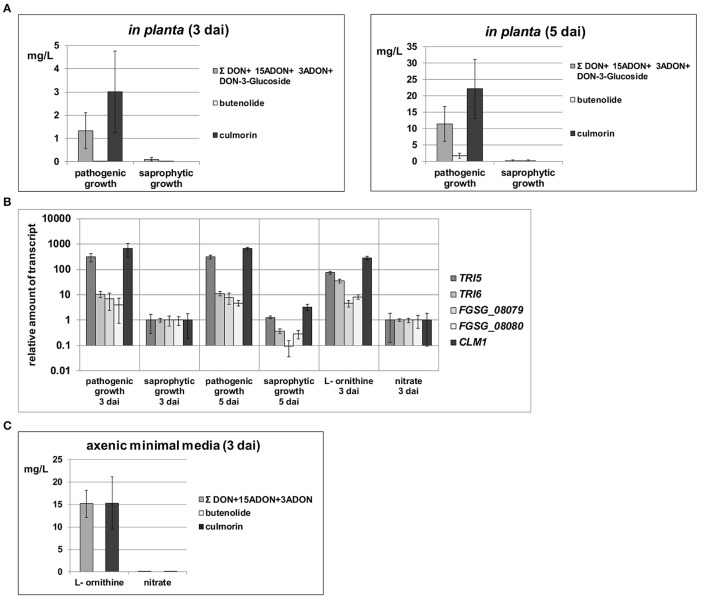
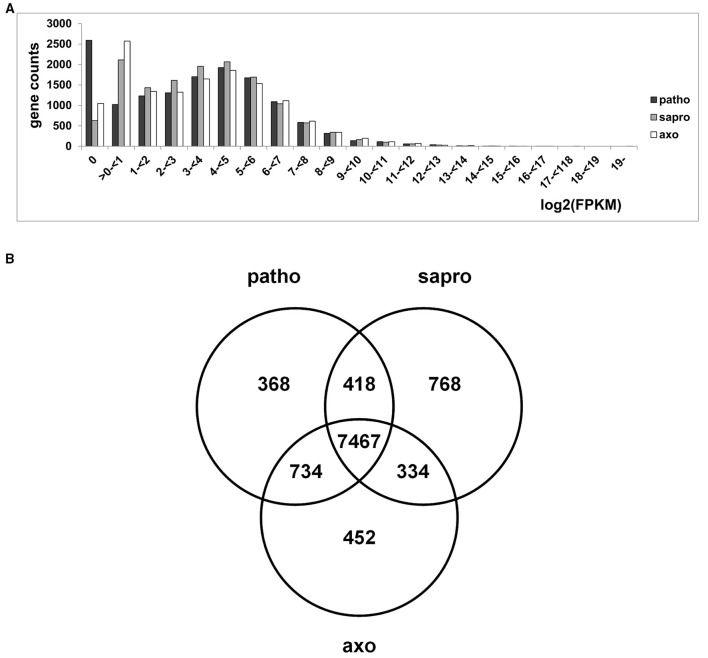
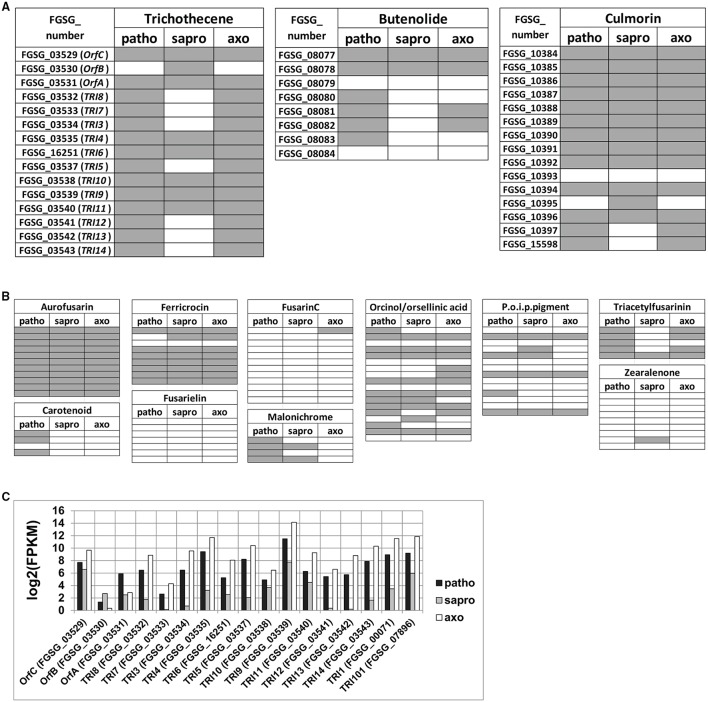
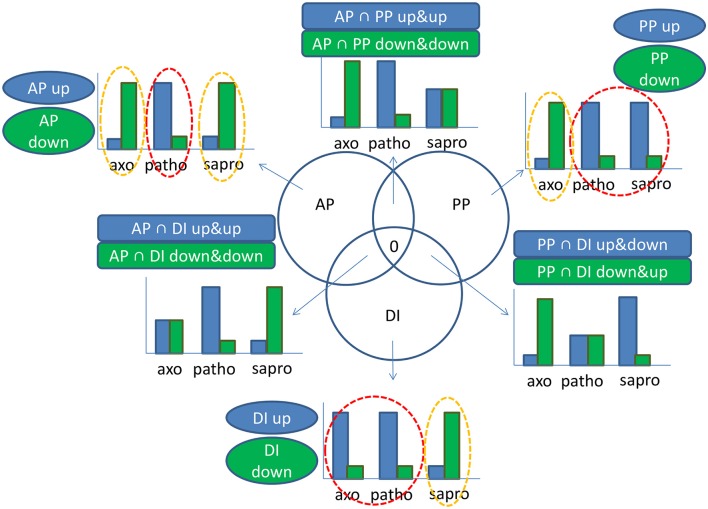
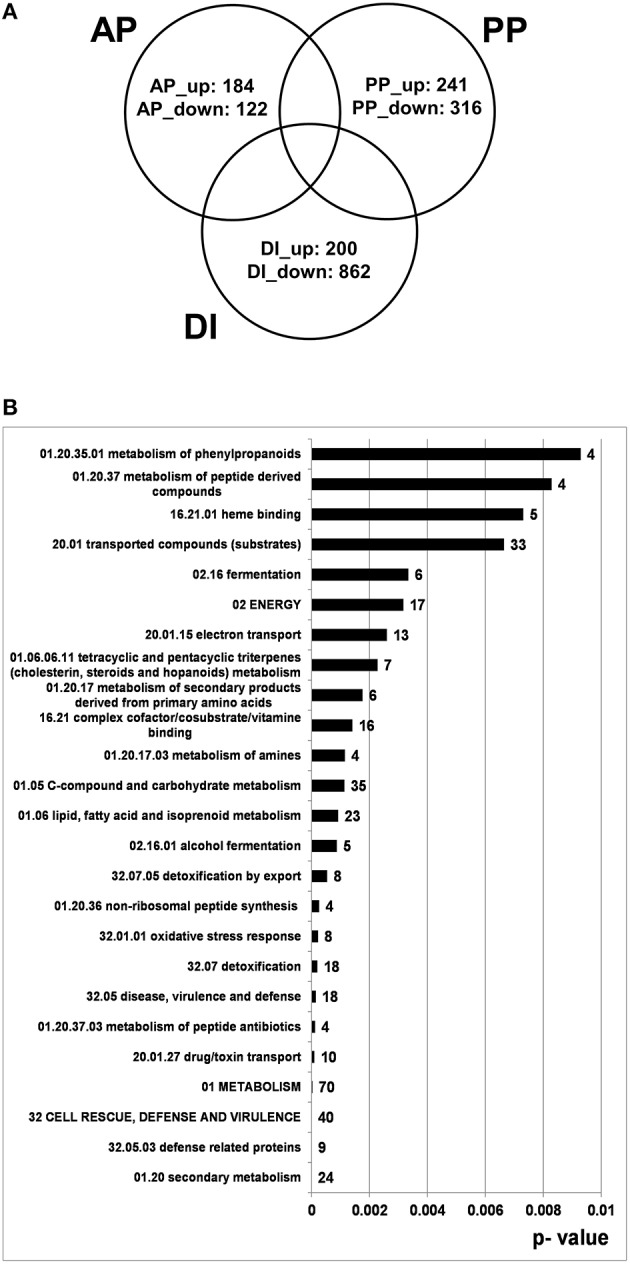
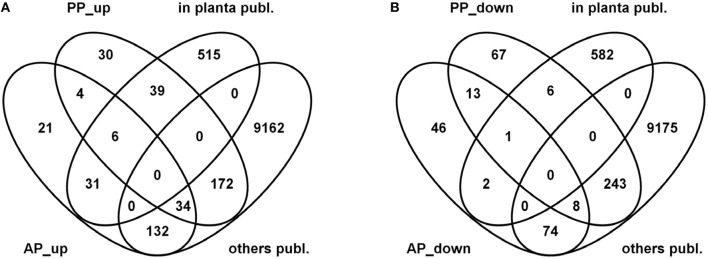
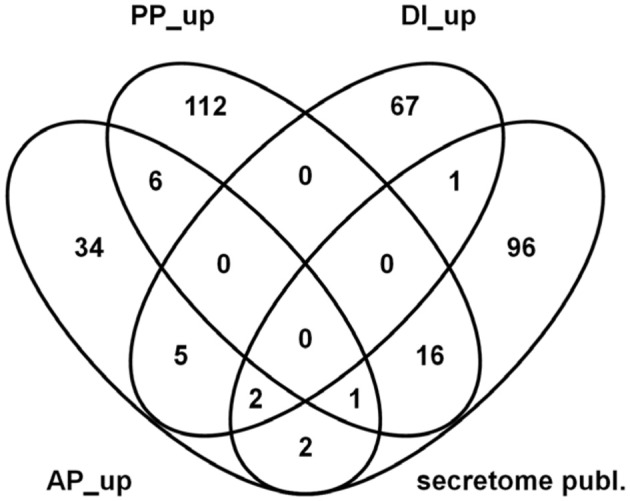
Fusarium graminearum is an opportunistic pathogen of cereals where it causes severe yield losses and concomitant mycotoxin contamination of the grains. The pathogen has mixed biotrophic and necrotrophic (saprophytic) growth phases during infection and the regulatory networks associated with these phases have so far always been analyzed together. In this study we compared the transcriptomes of fungal cells infecting a living, actively defending plant representing the mixed live style (pathogenic growth on living flowering wheat heads) to the response of the fungus infecting identical, but dead plant tissues (cold-killed flowering wheat heads) representing strictly saprophytic conditions. We found that the living plant actively suppressed fungal growth and promoted much higher toxin production in comparison to the identical plant tissue without metabolism suggesting that molecules signaling secondary metabolite induction are not pre-existing or not stable in the plant in sufficient amounts before infection. Differential gene expression analysis was used to define gene sets responding to the active or the passive plant as main impact factor and driver for gene expression. We correlated our results to the published F. graminearum transcriptomes, proteomes, and secretomes and found that only a limited number of in planta- expressed genes require the living plant for induction but the majority uses simply the plant tissue as signal. Many secondary metabolite (SM) gene clusters show a heterogeneous expression pattern within the cluster indicating that different genetic or epigenetic signals govern the expression of individual genes within a physically linked cluster. Our bioinformatic approach also identified fungal genes which were actively repressed by signals derived from the active plant and may thus represent direct targets of the plant defense against the invading pathogen.
Keywords: Fusarium; active plant; defense genes; passive plant; pathogenicity factors; secondary metabolism.
Figures
Similar articles
-
Comparative transcriptomics identifies the key in planta-expressed genes of Fusarium graminearum during infection of wheat varieties.Front Genet. 2023 Apr 18;14:1166832. doi: 10.3389/fgene.2023.1166832. eCollection 2023. Front Genet. 2023. PMID: 37144121 Free PMC article.
-
Priming of wheat with the green leaf volatile Z-3-hexenyl acetate enhances defense against Fusarium graminearum but boosts deoxynivalenol production.Plant Physiol. 2015 Apr;167(4):1671-84. doi: 10.1104/pp.15.00107. Epub 2015 Feb 20. Plant Physiol. 2015. PMID: 25713338 Free PMC article.
-
Systemic growth of F. graminearum in wheat plants and related accumulation of deoxynivalenol.Toxins (Basel). 2014 Apr 10;6(4):1308-24. doi: 10.3390/toxins6041308. Toxins (Basel). 2014. PMID: 24727554 Free PMC article.
-
Transcriptomics of cereal-Fusarium graminearum interactions: what we have learned so far.Mol Plant Pathol. 2018 Mar;19(3):764-778. doi: 10.1111/mpp.12561. Epub 2017 Jun 7. Mol Plant Pathol. 2018. PMID: 28411402 Free PMC article. Review.
-
Surfactin inhibits Fusarium graminearum by accumulating intracellular ROS and inducing apoptosis mechanisms.World J Microbiol Biotechnol. 2023 Oct 12;39(12):340. doi: 10.1007/s11274-023-03790-2. World J Microbiol Biotechnol. 2023. PMID: 37821760 Review.
Cited by
-
Functional Analysis of Genes Specifically Expressed during Aerial Hyphae Collapse as a Potential Signal for Perithecium Formation Induction in Fusarium graminearum.Plant Pathol J. 2024 Feb;40(1):83-97. doi: 10.5423/PPJ.FT.01.2024.0007. Epub 2024 Feb 1. Plant Pathol J. 2024. PMID: 38326961 Free PMC article.
-
Weighted gene co-expression network analysis unveils gene networks associated with the Fusarium head blight resistance in tetraploid wheat.BMC Genomics. 2019 Dec 3;20(1):925. doi: 10.1186/s12864-019-6161-8. BMC Genomics. 2019. PMID: 31795948 Free PMC article.
-
Comparative transcriptomics identifies the key in planta-expressed genes of Fusarium graminearum during infection of wheat varieties.Front Genet. 2023 Apr 18;14:1166832. doi: 10.3389/fgene.2023.1166832. eCollection 2023. Front Genet. 2023. PMID: 37144121 Free PMC article.
-
Genetic response to nitrogen starvation in the aggressive Eucalyptus foliar pathogen Teratosphaeria destructans.Curr Genet. 2021 Dec;67(6):981-990. doi: 10.1007/s00294-021-01208-w. Epub 2021 Aug 25. Curr Genet. 2021. PMID: 34432124
-
Two distinct Fusarium graminearum populations colonized European wheat in the past two decades.PLoS One. 2023 Dec 28;18(12):e0296302. doi: 10.1371/journal.pone.0296302. eCollection 2023. PLoS One. 2023. PMID: 38153926 Free PMC article.
References
-
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Methodol. 57, 289–300.
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases