

Changes in the Structure of the Microbial Community Associated with Nannochloropsis salina following Treatments with Antibiotics and Bioactive Compounds
- PMID: 27507966
- PMCID: PMC4960269
- DOI: 10.3389/fmicb.2016.01155
Changes in the Structure of the Microbial Community Associated with Nannochloropsis salina following Treatments with Antibiotics and Bioactive Compounds
Abstract
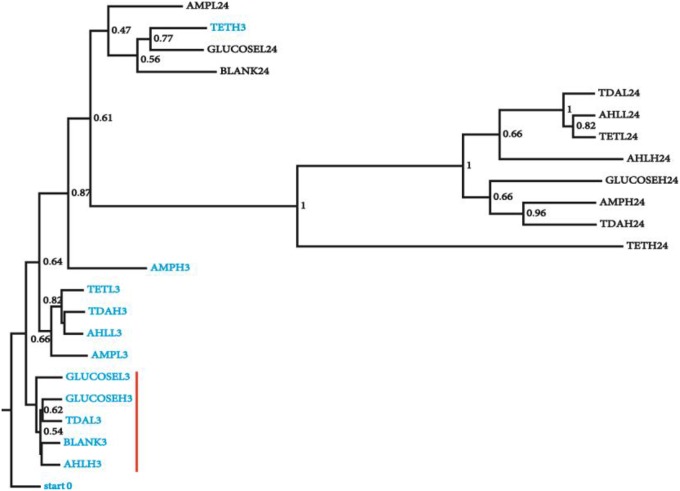
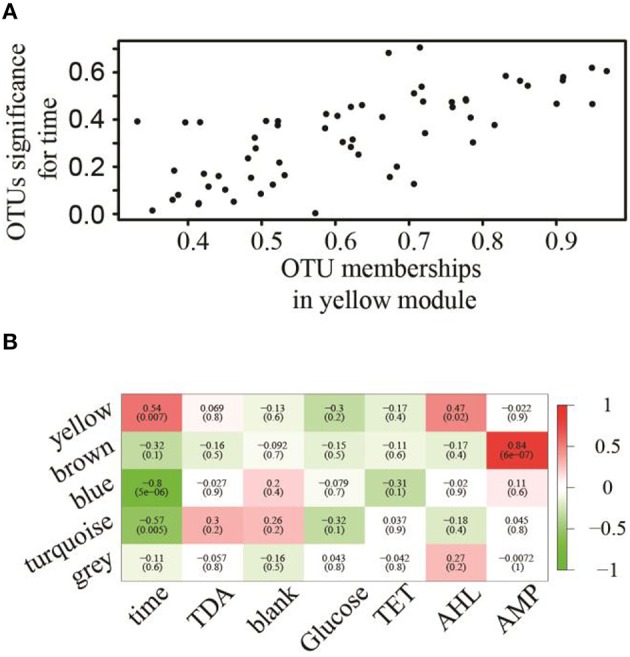
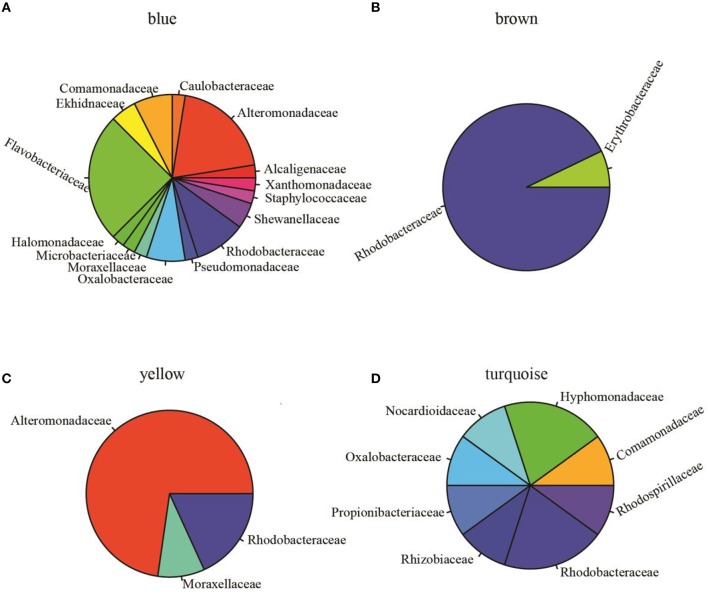
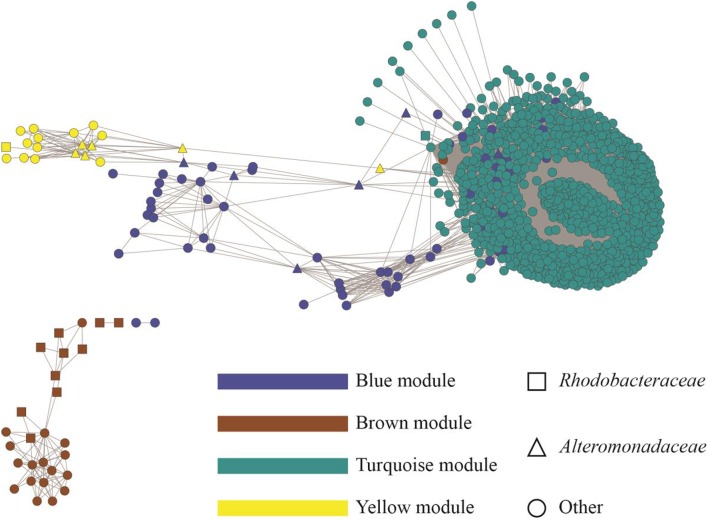
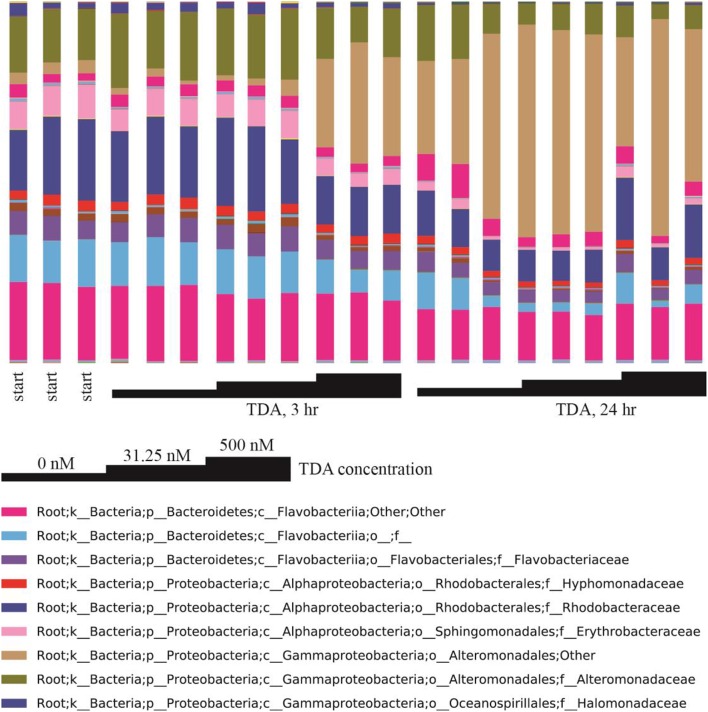
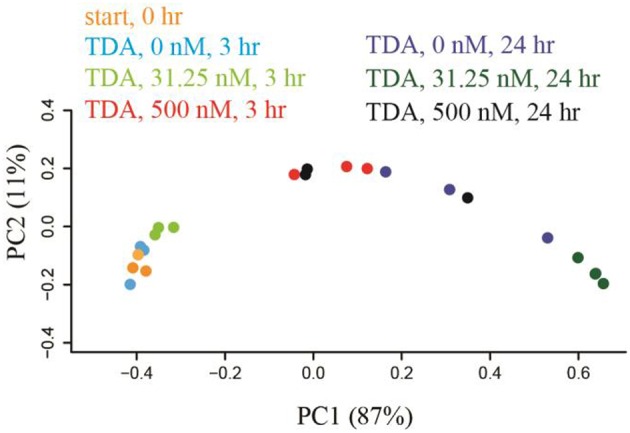
Open microalgae cultures host a myriad of bacteria, creating a complex system of interacting species that influence algal growth and health. Many algal microbiota studies have been conducted to determine the relative importance of bacterial taxa to algal culture health and physiological states, but these studies have not characterized the interspecies relationships in the microbial communities. We subjected Nanochroloropsis salina cultures to multiple chemical treatments (antibiotics and quorum sensing compounds) and obtained dense time-series data on changes to the microbial community using 16S gene amplicon metagenomic sequencing (21,029,577 reads for 23 samples) to measure microbial taxa-taxa abundance correlations. Short-term treatment with antibiotics resulted in substantially larger shifts in the microbiota structure compared to changes observed following treatment with signaling compounds and glucose. We also calculated operational taxonomic unit (OTU) associations and generated OTU correlation networks to provide an overview of possible bacterial OTU interactions. This analysis identified five major cohesive modules of microbiota with similar co-abundance profiles across different chemical treatments. The Eigengenes of OTU modules were examined for correlation with different external treatment factors. This correlation-based analysis revealed that culture age (time) and treatment types have primary effects on forming network modules and shaping the community structure. Additional network analysis detected Alteromonadeles and Alphaproteobacteria as having the highest centrality, suggesting these species are "keystone" OTUs in the microbial community. Furthermore, we illustrated that the chemical tropodithietic acid, which is secreted by several species in the Alphaproteobacteria taxon, is able to drastically change the structure of the microbiota within 3 h. Taken together, these results provide valuable insights into the structure of the microbiota associated with N. salina cultures and how these structures change in response to chemical perturbations.
Keywords: algae; correlation network; microbiota.
Figures
Similar articles
-
Longitudinal Analysis of Microbiota in Microalga Nannochloropsis salina Cultures.Microb Ecol. 2016 Jul;72(1):14-24. doi: 10.1007/s00248-016-0746-4. Epub 2016 Mar 8. Microb Ecol. 2016. PMID: 26956183
-
Bacterial Community Assembly, Succession, and Metabolic Function during Outdoor Cultivation of Microchloropsis salina.mSphere. 2022 Aug 31;7(4):e0023122. doi: 10.1128/msphere.00231-22. Epub 2022 Jun 22. mSphere. 2022. PMID: 35730934 Free PMC article.
-
Microbial Community Succession and Nutrient Cycling Responses following Perturbations of Experimental Saltwater Aquaria.mSphere. 2019 Feb 20;4(1):e00043-19. doi: 10.1128/mSphere.00043-19. mSphere. 2019. PMID: 30787117 Free PMC article.
-
Effects of host species and environment on the skin microbiome of Plethodontid salamanders.J Anim Ecol. 2018 Mar;87(2):341-353. doi: 10.1111/1365-2656.12726. Epub 2017 Aug 21. J Anim Ecol. 2018. PMID: 28682480
-
Temporal and spatial influences incur reconfiguration of Arctic heathland soil bacterial community structure.Environ Microbiol. 2016 Jun;18(6):1942-53. doi: 10.1111/1462-2920.13017. Epub 2015 Sep 16. Environ Microbiol. 2016. PMID: 26259508
Cited by
-
Effectiveness of cyclic treatment of municipal wastewater by Tetradesmus obliquus - Loofah biofilm, its internal community changes and potential for resource utilization.Water Res X. 2024 Aug 29;24:100254. doi: 10.1016/j.wroa.2024.100254. eCollection 2024 Sep 1. Water Res X. 2024. PMID: 39281025 Free PMC article.
-
Assembly of a Benthic Microbial Community in a Eutrophic Bay with a Long History of Oyster Culturing.Microorganisms. 2021 Sep 24;9(10):2019. doi: 10.3390/microorganisms9102019. Microorganisms. 2021. PMID: 34683340 Free PMC article.
-
Bacterial Operational Taxonomic Units Replace the Interactive Roles of Other Operational Taxonomic Units Under Strong Environmental Changes.Curr Genomics. 2020 Nov;21(7):512-524. doi: 10.2174/1389202921999200716104355. Curr Genomics. 2020. PMID: 33214767 Free PMC article.
-
Tropodithietic Acid, a Multifunctional Antimicrobial, Facilitates Adaption and Colonization of the Producer, Phaeobacter piscinae.mSphere. 2023 Feb 21;8(1):e0051722. doi: 10.1128/msphere.00517-22. Epub 2023 Jan 9. mSphere. 2023. PMID: 36622251 Free PMC article.
-
Guild-based analysis for understanding gut microbiome in human health and diseases.Genome Med. 2021 Feb 9;13(1):22. doi: 10.1186/s13073-021-00840-y. Genome Med. 2021. PMID: 33563315 Free PMC article.
References
-
- Bartram A. K., Lynch M. D. J., Stearns J. C., Moreno-Hagelsieb G., Neufeld J. D. (2011). Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl. Environ. Microbiol. 77, 3846–3852. 10.1128/AEM.02772-10 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources