

From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline
- PMID: 27508061
- PMCID: PMC4934518
- DOI: 10.12688/f1000research.8987.2
From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline
Abstract
In recent years, RNA sequencing (RNA-seq) has become a very widely used technology for profiling gene expression. One of the most common aims of RNA-seq profiling is to identify genes or molecular pathways that are differentially expressed (DE) between two or more biological conditions. This article demonstrates a computational workflow for the detection of DE genes and pathways from RNA-seq data by providing a complete analysis of an RNA-seq experiment profiling epithelial cell subsets in the mouse mammary gland. The workflow uses R software packages from the open-source Bioconductor project and covers all steps of the analysis pipeline, including alignment of read sequences, data exploration, differential expression analysis, visualization and pathway analysis. Read alignment and count quantification is conducted using the Rsubread package and the statistical analyses are performed using the edgeR package. The differential expression analysis uses the quasi-likelihood functionality of edgeR.
Keywords: R software; RNA sequencing; gene expression; molecular pathways.
Conflict of interest statement
Figures
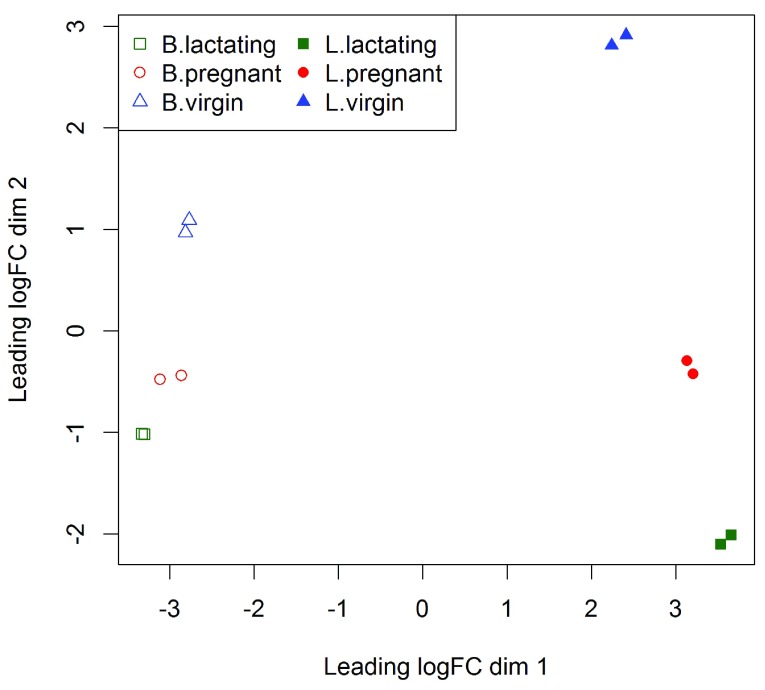
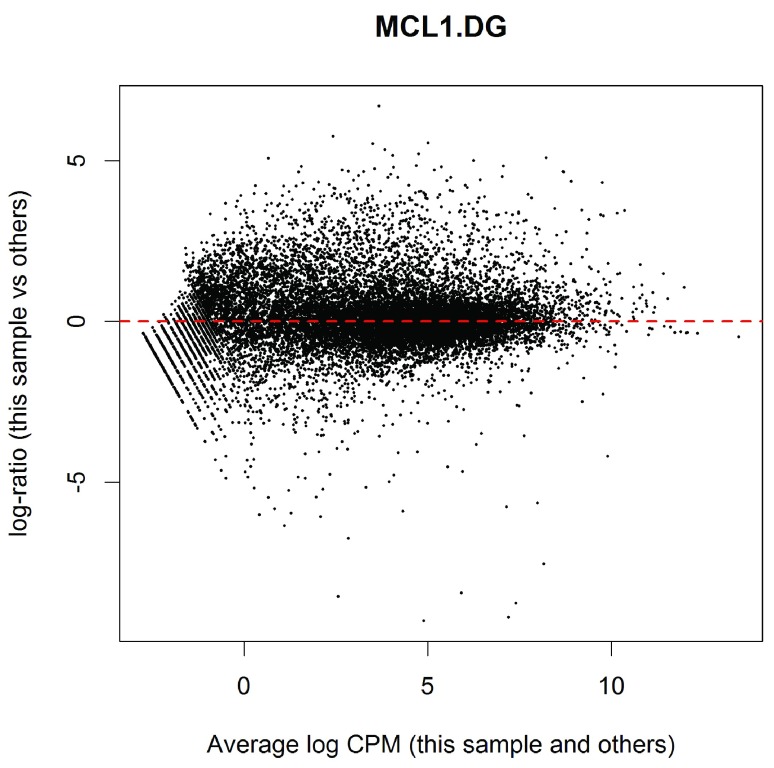
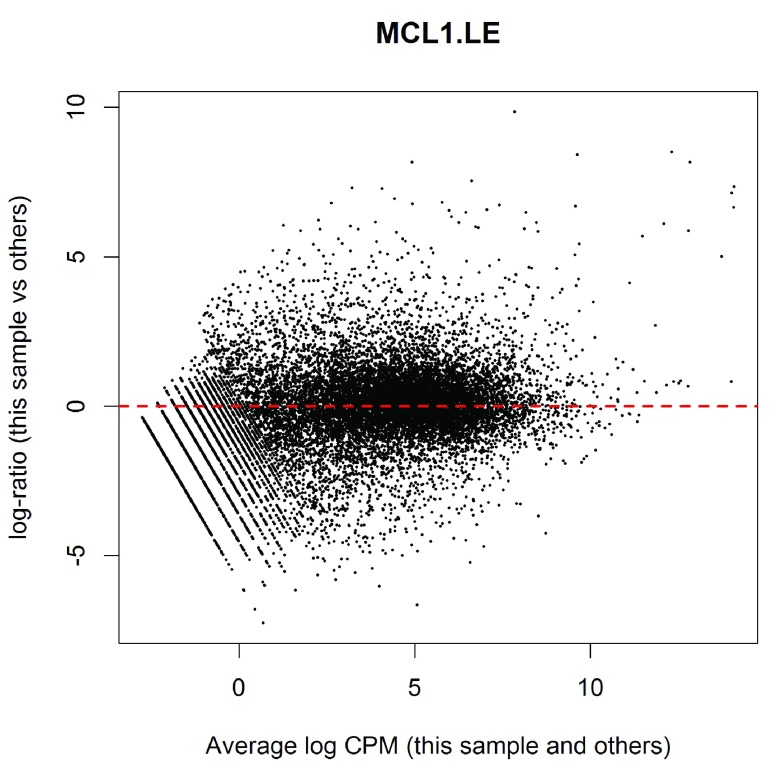
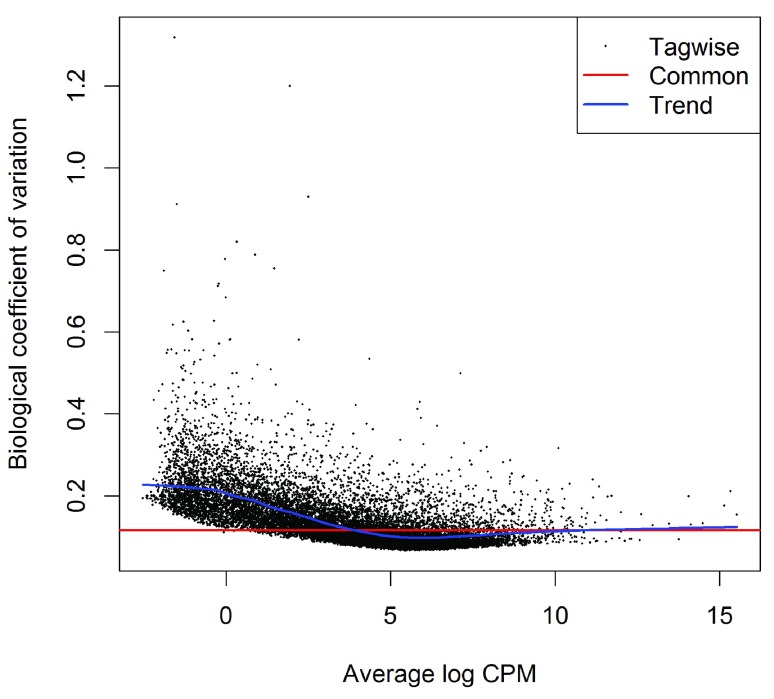
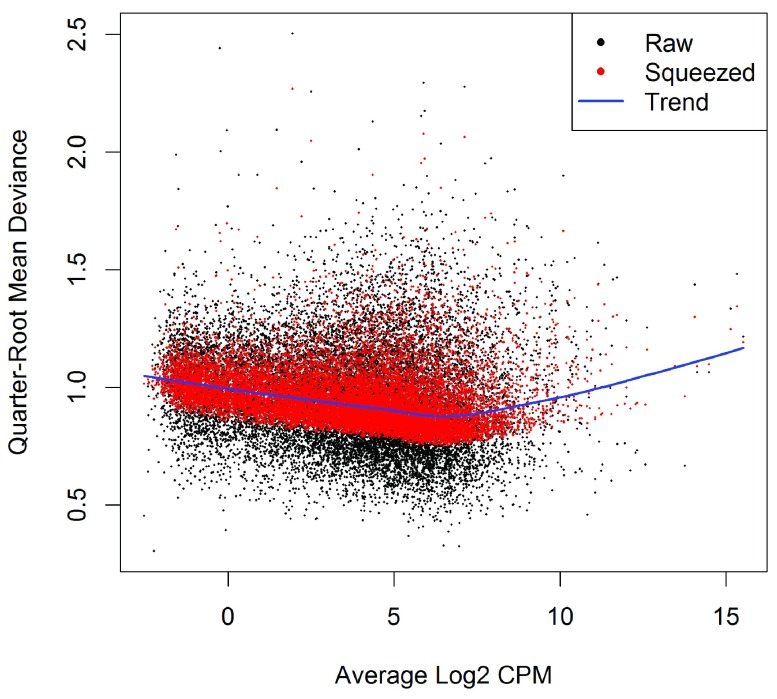
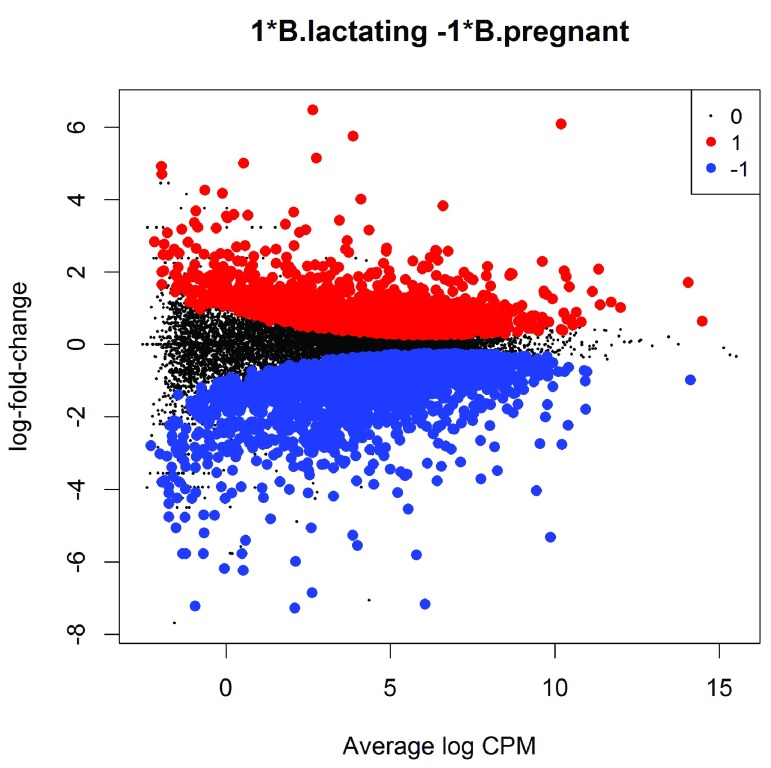
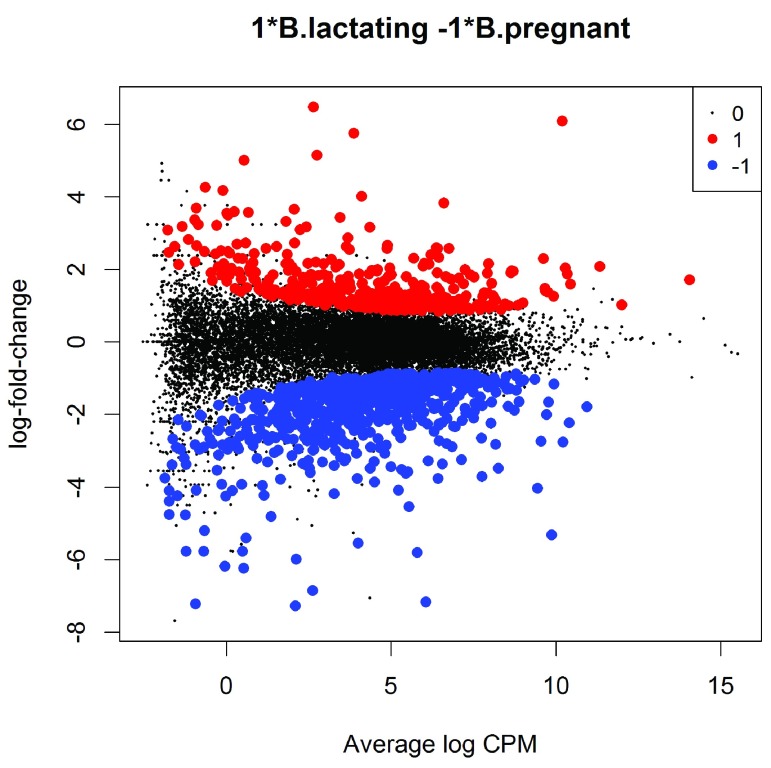
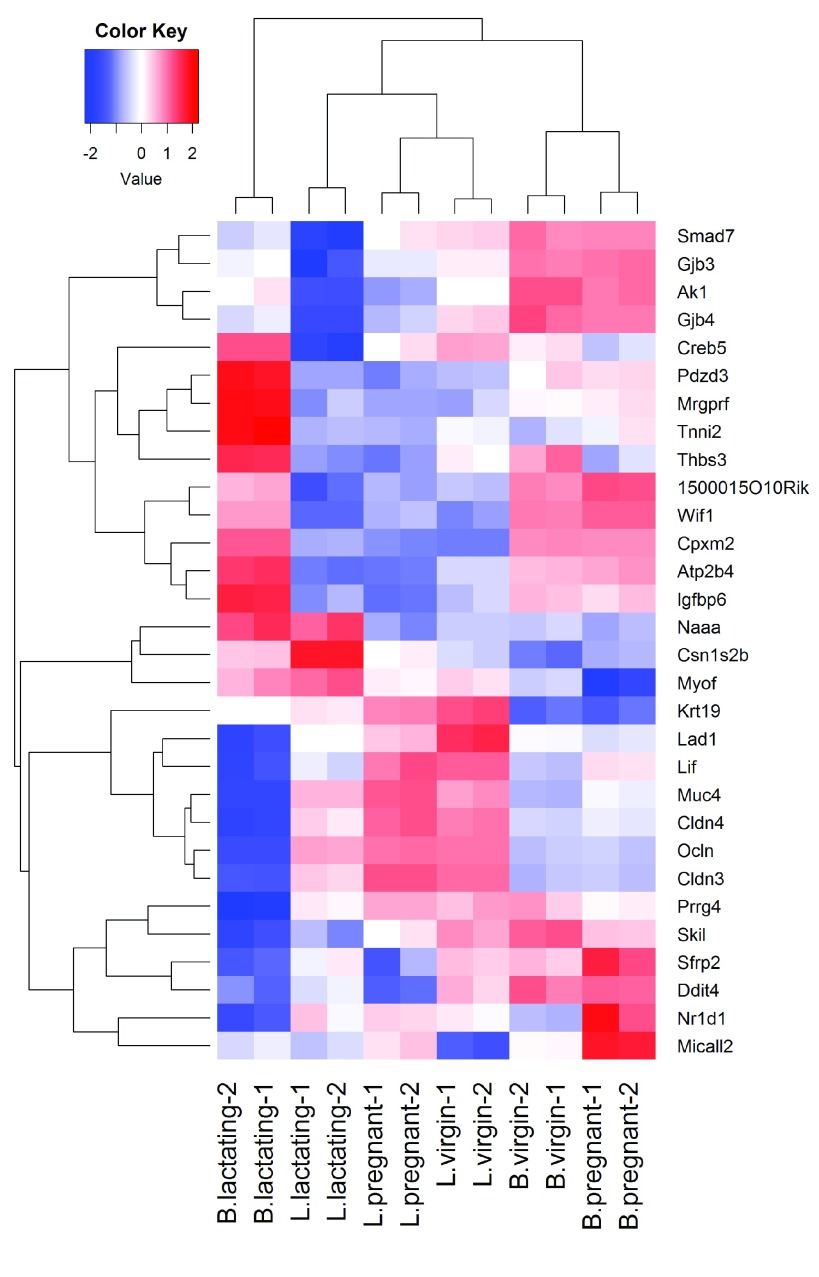
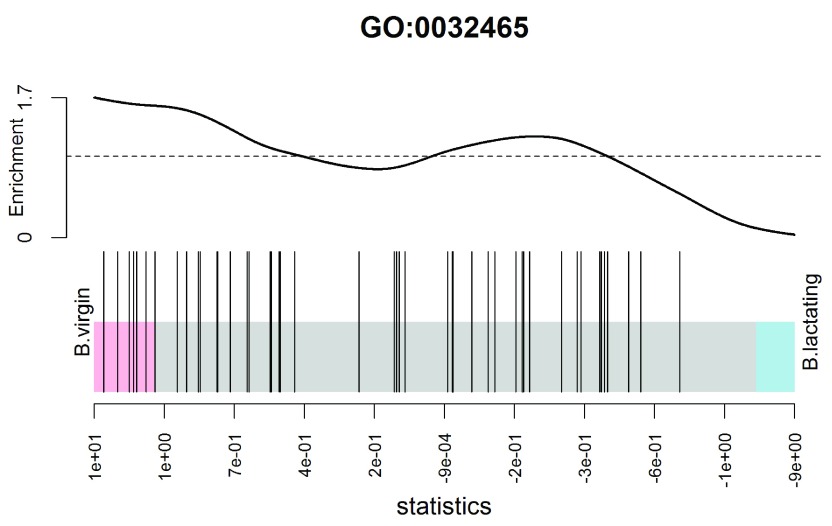
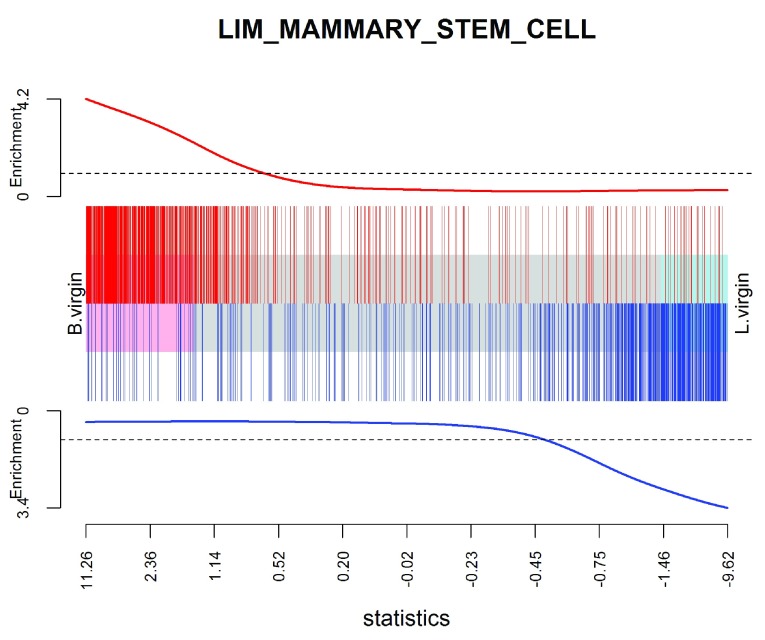
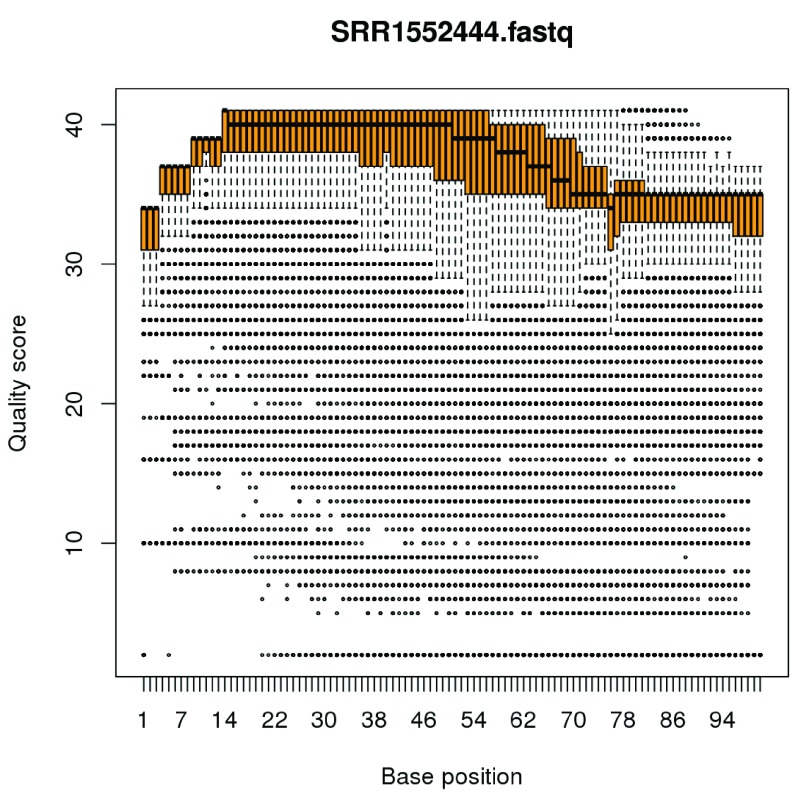
References
LinkOut - more resources
Full Text Sources
Other Literature Sources