

Intracellular calcium increases in vascular smooth muscle cells with progression of chronic kidney disease in a rat model
- PMID: 27510531
- PMCID: PMC5837609
- DOI: 10.1093/ndt/gfw274
Intracellular calcium increases in vascular smooth muscle cells with progression of chronic kidney disease in a rat model
Abstract
Background: Vascular smooth muscle cells (VSMCs) exhibit phenotypic plasticity, promoting vascular calcification and increasing cardiovascular risk. Changes in VSMC intracellular calcium ([Ca 2+ ] i ) are a major determinant of plasticity, but little is known about changes in [Ca 2+ ] i in chronic kidney disease (CKD). We have previously demonstrated such plasticity in aortas from our rat model of CKD and therefore sought to examine changes in [Ca 2+ ] i during CKD progression.
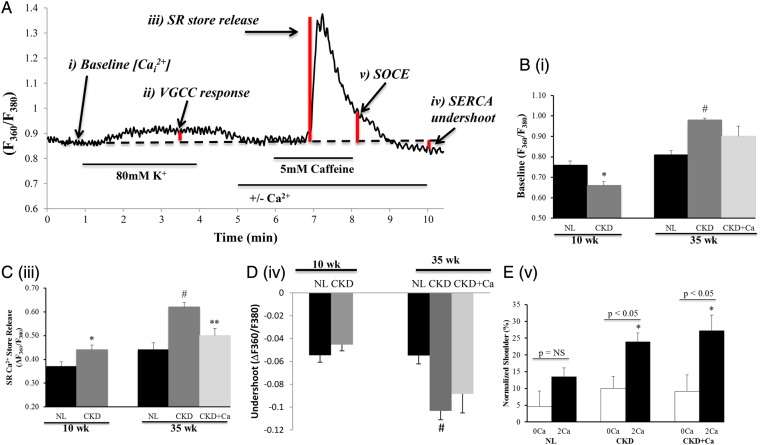
Materials and methods: We examined freshly isolated VSMCs from aortas of normal rats, Cy/+ rats (CKD) with early and advanced CKD, and advanced CKD rats treated without and with 3% calcium gluconate (CKD + Ca 2+ ) to lower parathyroid hormone (PTH) levels. [Ca 2+ ] i was measured with fura-2.
Results: Cy/+ rats developed progressive CKD, as assessed by plasma levels of blood urea nitrogen, calcium, phosphorus, parathyroid hormone and fibroblast growth factor 23. VSMCs isolated from rats with CKD demonstrated biphasic alterations in resting [Ca 2+ ] i : VSMCs from rats with early CKD exhibited reduced resting [Ca 2+ ] i , while VSMCs from rats with advanced CKD exhibited elevated resting [Ca 2+ ] i . Caffeine-induced sarcoplasmic reticulum (SR) Ca 2+ store release was modestly increased in early CKD and was more drastically increased in advanced CKD. The advanced CKD elevation in SR Ca 2+ store release was associated with a significant increase in the activity of the sarco-endoplasmic reticulum Ca 2+ ATPase (SERCA); however, SERCA2a protein expression was decreased in advanced CKD. Following SR Ca 2+ store release, recovery of [Ca 2+ ] i in the presence of caffeine and extracellular Ca 2+ was attenuated in VSMCs from rats with advanced CKD. This impairment, together with reductions in expression of the Na + /Ca 2+ exchanger, suggest a reduction in Ca 2+ extrusion capability. Finally, store-operated Ca 2+ entry (SOCE) was assessed following SR Ca 2+ store depletion. Ca 2+ entry during recovery from caffeine-induced SR Ca 2+ store release was elevated in advanced CKD, suggesting a role for exacerbated SOCE with progressing CKD.
Conclusions: With progressive CKD in the Cy/+ rat there is increased resting [Ca 2+ ] i in VSMCs due, in part, to increased SOCE and impaired calcium extrusion from the cell. Such changes may predispose VSMCs to phenotypic changes that are a prerequisite to calcification.
Keywords: calcium signaling; cell phenotype; chronic kidney disease; rat model; vascular smooth muscle cells.
Published by Oxford University Press on behalf of ERA-EDTA 2016. This work is written by US Government employees and is in the public domain in the US.
Figures



Similar articles
-
Phosphate-induced ORAI1 expression and store-operated Ca2+ entry in aortic smooth muscle cells.J Mol Med (Berl). 2019 Oct;97(10):1465-1475. doi: 10.1007/s00109-019-01824-7. Epub 2019 Aug 5. J Mol Med (Berl). 2019. PMID: 31385016
-
Ca2+ handling is altered when arterial myocytes progress from a contractile to a proliferative phenotype in culture.Am J Physiol Cell Physiol. 2008 Sep;295(3):C779-90. doi: 10.1152/ajpcell.00173.2008. Epub 2008 Jul 2. Am J Physiol Cell Physiol. 2008. PMID: 18596214 Free PMC article.
-
Defective autophagy in vascular smooth muscle cells alters contractility and Ca²⁺ homeostasis in mice.Am J Physiol Heart Circ Physiol. 2015 Mar 15;308(6):H557-67. doi: 10.1152/ajpheart.00659.2014. Epub 2015 Jan 9. Am J Physiol Heart Circ Physiol. 2015. PMID: 25576626
-
Signaling pathways involved in vascular smooth muscle cell calcification during hyperphosphatemia.Cell Mol Life Sci. 2019 Jun;76(11):2077-2091. doi: 10.1007/s00018-019-03054-z. Epub 2019 Mar 18. Cell Mol Life Sci. 2019. PMID: 30887097 Free PMC article. Review.
-
Calcium Signaling Dynamics in Vascular Cells and Their Dysregulation in Vascular Disease.Biomolecules. 2025 Jun 18;15(6):892. doi: 10.3390/biom15060892. Biomolecules. 2025. PMID: 40563532 Free PMC article. Review.
Cited by
-
Protection Effect of Exogenous Fibroblast Growth Factor 21 on the Kidney Injury in Vascular Calcification Rats.Chin Med J (Engl). 2018 Mar 5;131(5):532-538. doi: 10.4103/0366-6999.226065. Chin Med J (Engl). 2018. PMID: 29483386 Free PMC article.
-
Oxidative Stress Related to Plasmalemmal and Mitochondrial Phosphate Transporters in Vascular Calcification.Antioxidants (Basel). 2022 Mar 2;11(3):494. doi: 10.3390/antiox11030494. Antioxidants (Basel). 2022. PMID: 35326144 Free PMC article. Review.
-
Dietary potassium regulates vascular calcification and arterial stiffness.JCI Insight. 2017 Oct 5;2(19):e94920. doi: 10.1172/jci.insight.94920. JCI Insight. 2017. PMID: 28978809 Free PMC article.
-
Skeletal and cardiovascular consequences of a positive calcium balance during hemodialysis.J Bras Nefrol. 2021 Oct-Dec;43(4):539-550. doi: 10.1590/2175-8239-JBN-2020-0098. J Bras Nefrol. 2021. PMID: 33107900 Free PMC article. Review.
-
The mechanosensitive Piezo1 channels contribute to the arterial medial calcification.Front Physiol. 2022 Nov 10;13:1037230. doi: 10.3389/fphys.2022.1037230. eCollection 2022. Front Physiol. 2022. PMID: 36439266 Free PMC article.
References
-
- Go AS, Chertow GM, Fan D et al. . Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004; 351: 1296–1305 - PubMed
-
- Herzog CA, Asinger RW, Berger AK et al. . Cardiovascular disease in chronic kidney disease. A clinical update from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2011; 80: 572–586 - PubMed
-
- Moe SM, Seifert MF, Chen NX et al. . R-568 reduces ectopic calcification in a rat model of chronic kidney disease-mineral bone disorder (CKD-MBD). Nephrol Dial Transplant 2009; 24: 2371–2377 - PubMed
-
- Moe SM, Drueke T, Lameire N et al. . Chronic kidney disease-mineral-bone disorder: a new paradigm. Adv Chronic Kidney Dis 2007; 14: 3–12 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
