

Inhibition of DNA Methyltransferases Blocks Mutant Huntingtin-Induced Neurotoxicity
- PMID: 27516062
- PMCID: PMC4981892
- DOI: 10.1038/srep31022
Inhibition of DNA Methyltransferases Blocks Mutant Huntingtin-Induced Neurotoxicity
Erratum in
-
Erratum: Inhibition of DNA Methyltransferases Blocks Mutant Huntingtin-Induced Neurotoxicity.Sci Rep. 2016 Sep 21;6:33766. doi: 10.1038/srep33766. Sci Rep. 2016. PMID: 27649847 Free PMC article. No abstract available.
Abstract
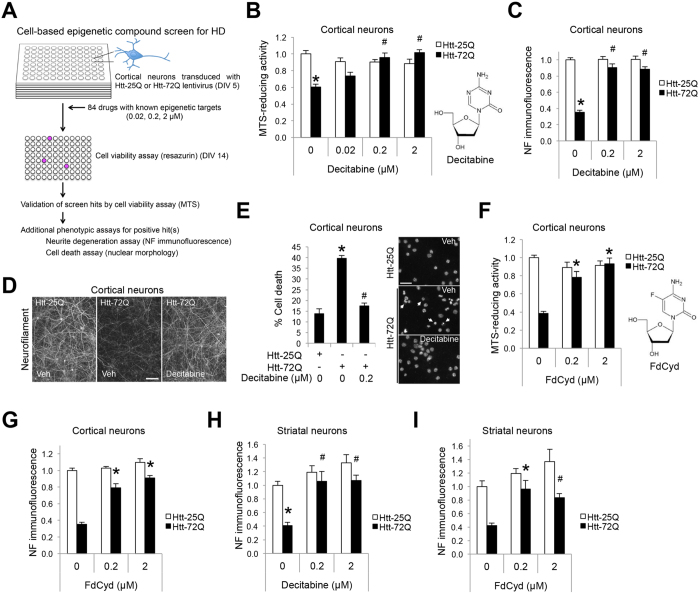
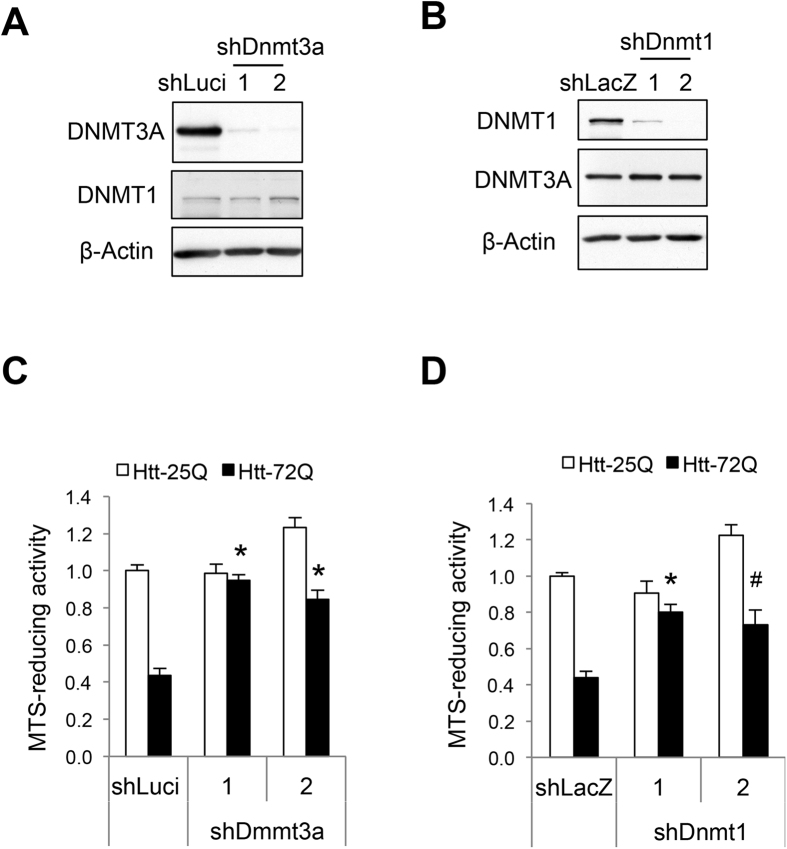
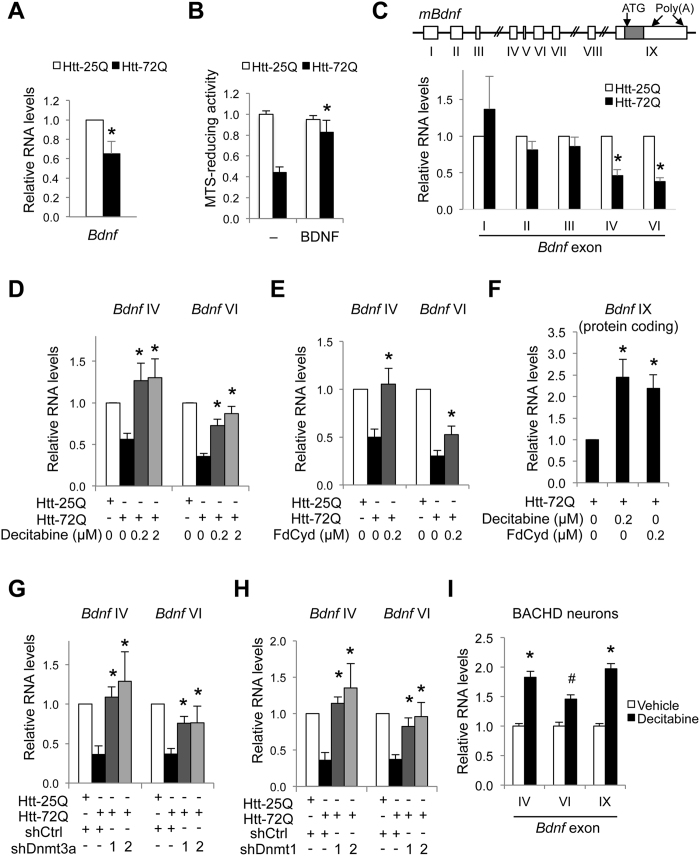
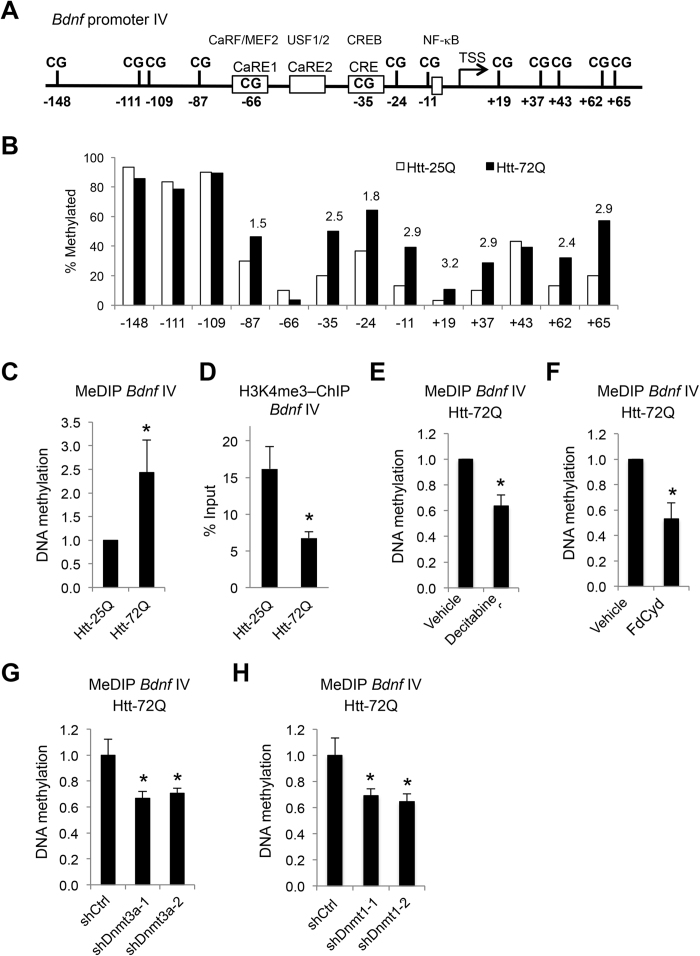
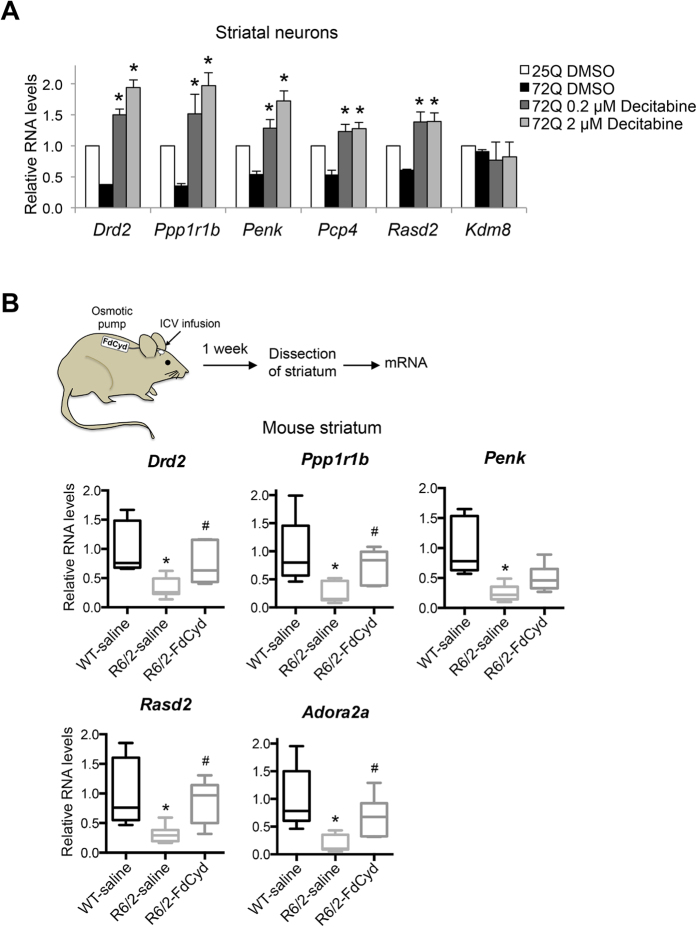
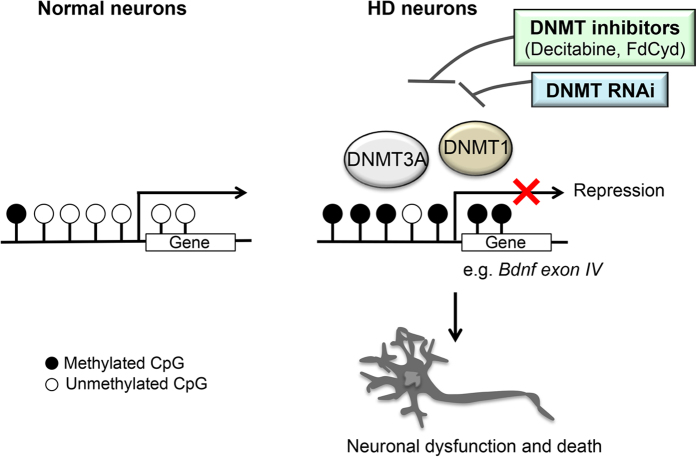
Although epigenetic abnormalities have been described in Huntington's disease (HD), the causal epigenetic mechanisms driving neurodegeneration in HD cortex and striatum remain undefined. Using an epigenetic pathway-targeted drug screen, we report that inhibitors of DNA methyltransferases (DNMTs), decitabine and FdCyd, block mutant huntingtin (Htt)-induced toxicity in primary cortical and striatal neurons. In addition, knockdown of DNMT3A or DNMT1 protected neurons against mutant Htt-induced toxicity, together demonstrating a requirement for DNMTs in mutant Htt-triggered neuronal death and suggesting a neurodegenerative mechanism based on DNA methylation-mediated transcriptional repression. Inhibition of DNMTs in HD model primary cortical or striatal neurons restored the expression of several key genes, including Bdnf, an important neurotrophic factor implicated in HD. Accordingly, the Bdnf promoter exhibited aberrant cytosine methylation in mutant Htt-expressing cortical neurons. In vivo, pharmacological inhibition of DNMTs in HD mouse brains restored the mRNA levels of key striatal genes known to be downregulated in HD. Thus, disturbances in DNA methylation play a critical role in mutant Htt-induced neuronal dysfunction and death, raising the possibility that epigenetic strategies targeting abnormal DNA methylation may have therapeutic utility in HD.
Figures
Similar articles
-
The role of Twist1 in mutant huntingtin-induced transcriptional alterations and neurotoxicity.J Biol Chem. 2018 Jul 27;293(30):11850-11866. doi: 10.1074/jbc.RA117.001211. Epub 2018 Jun 11. J Biol Chem. 2018. PMID: 29891550 Free PMC article.
-
PRMT5- mediated symmetric arginine dimethylation is attenuated by mutant huntingtin and is impaired in Huntington's disease (HD).Cell Cycle. 2015;14(11):1716-29. doi: 10.1080/15384101.2015.1033595. Cell Cycle. 2015. PMID: 25927346 Free PMC article.
-
Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington's disease.Cell Death Differ. 2004 Apr;11(4):424-38. doi: 10.1038/sj.cdd.4401358. Cell Death Differ. 2004. PMID: 14713958
-
Epigenetic regulation in Huntington's disease.Neurochem Int. 2021 Sep;148:105074. doi: 10.1016/j.neuint.2021.105074. Epub 2021 May 24. Neurochem Int. 2021. PMID: 34038804 Free PMC article. Review.
-
Selective degeneration in YAC mouse models of Huntington disease.Brain Res Bull. 2007 Apr 30;72(2-3):124-31. doi: 10.1016/j.brainresbull.2006.10.018. Epub 2006 Nov 16. Brain Res Bull. 2007. PMID: 17352936 Review.
Cited by
-
Role of DNMTs in the Brain.Adv Exp Med Biol. 2022;1389:363-394. doi: 10.1007/978-3-031-11454-0_15. Adv Exp Med Biol. 2022. PMID: 36350518 Review.
-
DNA Methyltransferase 1 (DNMT1) Acts on Neurodegeneration by Modulating Proteostasis-Relevant Intracellular Processes.Int J Mol Sci. 2020 Jul 30;21(15):5420. doi: 10.3390/ijms21155420. Int J Mol Sci. 2020. PMID: 32751461 Free PMC article.
-
Dynamic Regulation of DNA Methylation and Brain Functions.Biology (Basel). 2023 Jan 18;12(2):152. doi: 10.3390/biology12020152. Biology (Basel). 2023. PMID: 36829430 Free PMC article. Review.
-
Brain-Derived Neurotrophic Factor Mediated Perfluorooctane Sulfonate Induced-Neurotoxicity via Epigenetics Regulation in SK-N-SH Cells.Int J Mol Sci. 2017 Apr 24;18(4):893. doi: 10.3390/ijms18040893. Int J Mol Sci. 2017. PMID: 28441774 Free PMC article.
-
The DNA Methylation in Neurological Diseases.Cells. 2022 Oct 31;11(21):3439. doi: 10.3390/cells11213439. Cells. 2022. PMID: 36359835 Free PMC article. Review.
References
-
- Walker F. O. Huntington’s disease. Lancet 369, 218–228 (2007). - PubMed
-
- Ross C. A. et al.. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nature reviews. Neurology 10, 204–216 (2014). - PubMed
-
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983 (1993). - PubMed
-
- Sugars K. L. & Rubinsztein D. C. Transcriptional abnormalities in Huntington disease. Trends Genet 19, 233–238 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
