

Of Inflammasomes and Alarmins: IL-1β and IL-1α in Kidney Disease
- PMID: 27516236
- PMCID: PMC5004665
- DOI: 10.1681/ASN.2016020177
Of Inflammasomes and Alarmins: IL-1β and IL-1α in Kidney Disease
Abstract
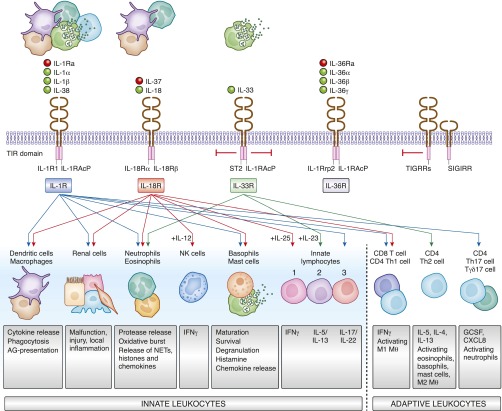
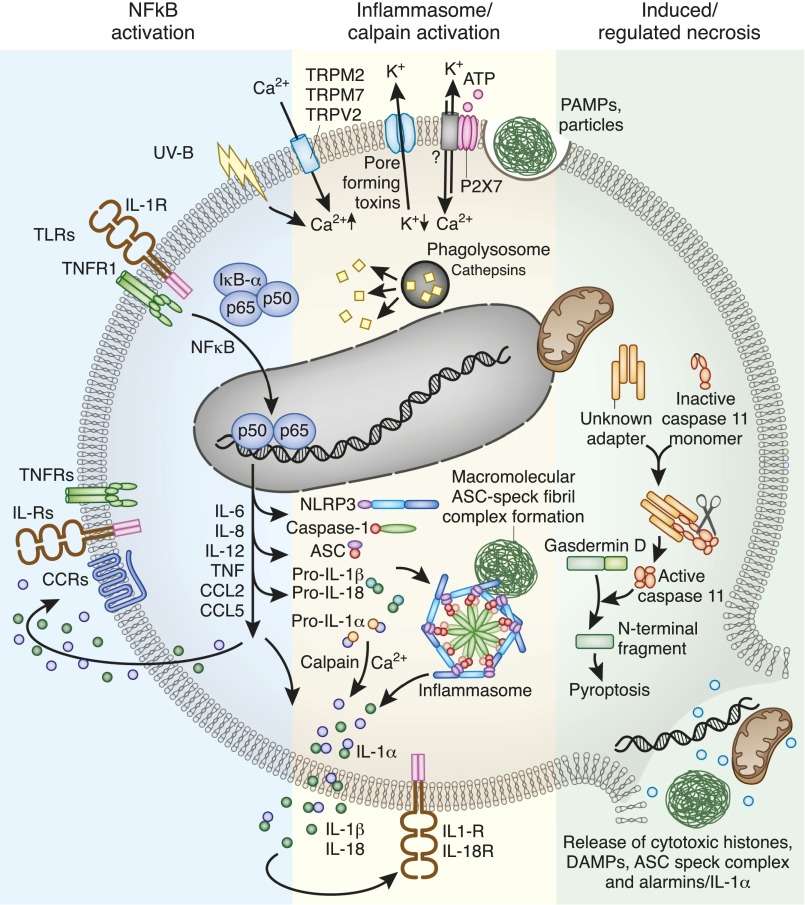
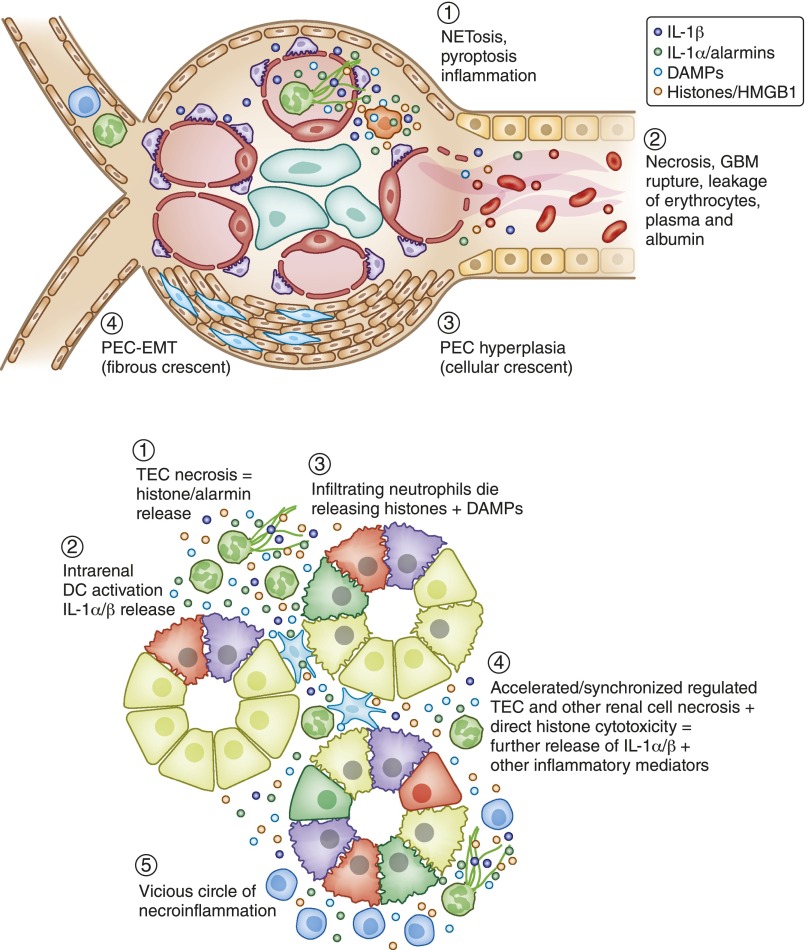
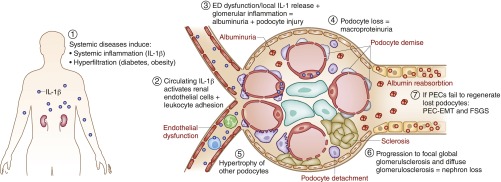
Kidney injury implies danger signaling and a response by the immune system. The inflammasome is a central danger recognition platform that triggers local and systemic inflammation. In immune cells, inflammasome activation causes the release of mature IL-1β and of the alarmin IL-1α Dying cells release IL-1α also, independently of the inflammasome. Both IL-1α and IL-1β ligate the same IL-1 receptor (IL-1R) that is present on nearly all cells inside and outside the kidney, further amplifying cytokine and chemokine release. Thus, the inflammasome-IL-1α/IL-β-IL-1R system is a central element of kidney inflammation and the systemic consequences. Seminal discoveries of recent years have expanded this central paradigm of inflammation. This review gives an overview of arising concepts of inflammasome and IL-1α/β regulation in renal cells and in experimental kidney disease models. There is a pipeline of compounds that can interfere with the inflammasome-IL-1α/IL-β-IL-1R system, ranging from recently described small molecule inhibitors of NLRP3, a component of the inflammasome complex, to regulatory agency-approved IL-1-neutralizing biologic drugs. Based on strong theoretic and experimental rationale, the potential therapeutic benefits of using such compounds to block the inflammasome-IL-1α/IL-β-IL-1R system in kidney disease should be further explored.
Keywords: acute kidney injury; chronic kidney disease; glomerulonephritis; inflammation; innate immunity.
Copyright © 2016 by the American Society of Nephrology.
Figures
References
-
- Anders HJ, Muruve DA: The inflammasomes in kidney disease. J Am Soc Nephrol 22: 1007–1018, 2011 - PubMed
-
- Lomedico PT, Gubler U, Hellmann CP, Dukovich M, Giri JG, Pan YC, Collier K, Semionow R, Chua AO, Mizel SB: Cloning and expression of murine interleukin-1 cDNA in Escherichia coli. Nature 312: 458–462, 1984 - PubMed
-
- Netea MG, van de Veerdonk FL, van der Meer JW, Dinarello CA, Joosten LA: Inflammasome-independent regulation of IL-1-family cytokines. Annu Rev Immunol 33: 49–77, 2015 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
