

Eosinophil peroxidase activates cells by HER2 receptor engagement and β1-integrin clustering with downstream MAPK cell signaling
- PMID: 27519953
- PMCID: PMC5070911
- DOI: 10.1016/j.clim.2016.08.009
Eosinophil peroxidase activates cells by HER2 receptor engagement and β1-integrin clustering with downstream MAPK cell signaling
Abstract
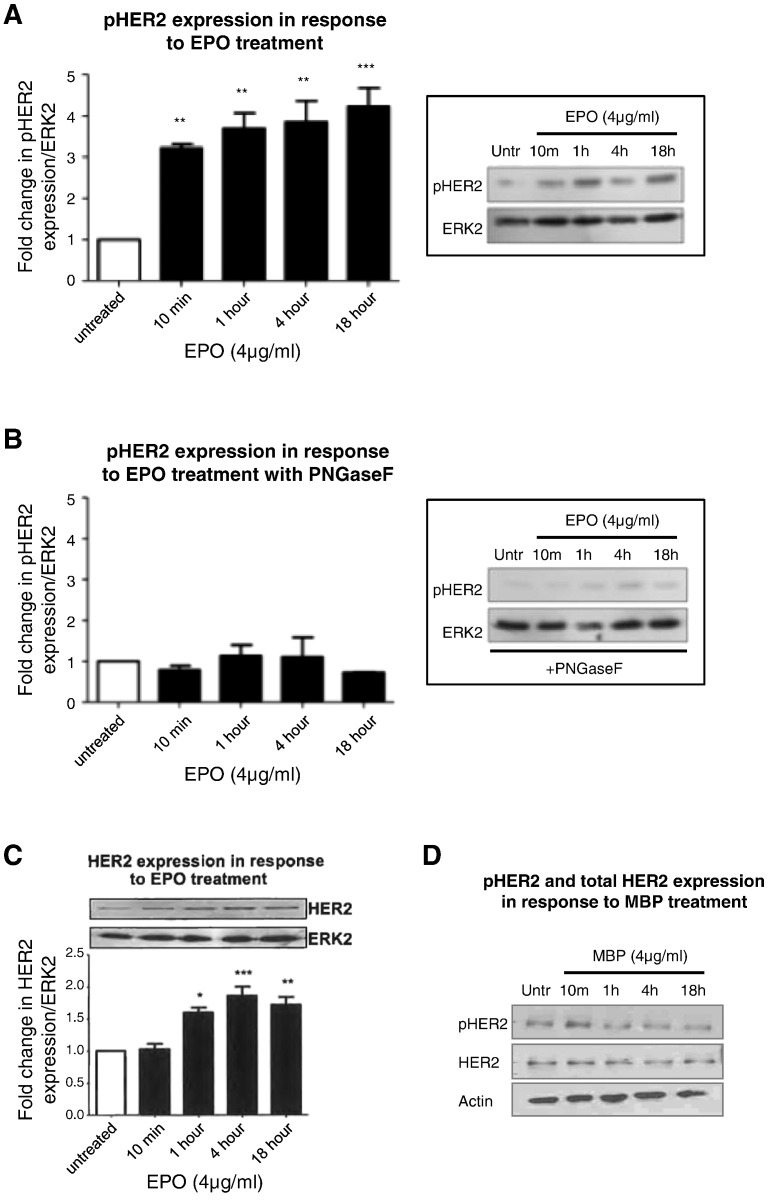
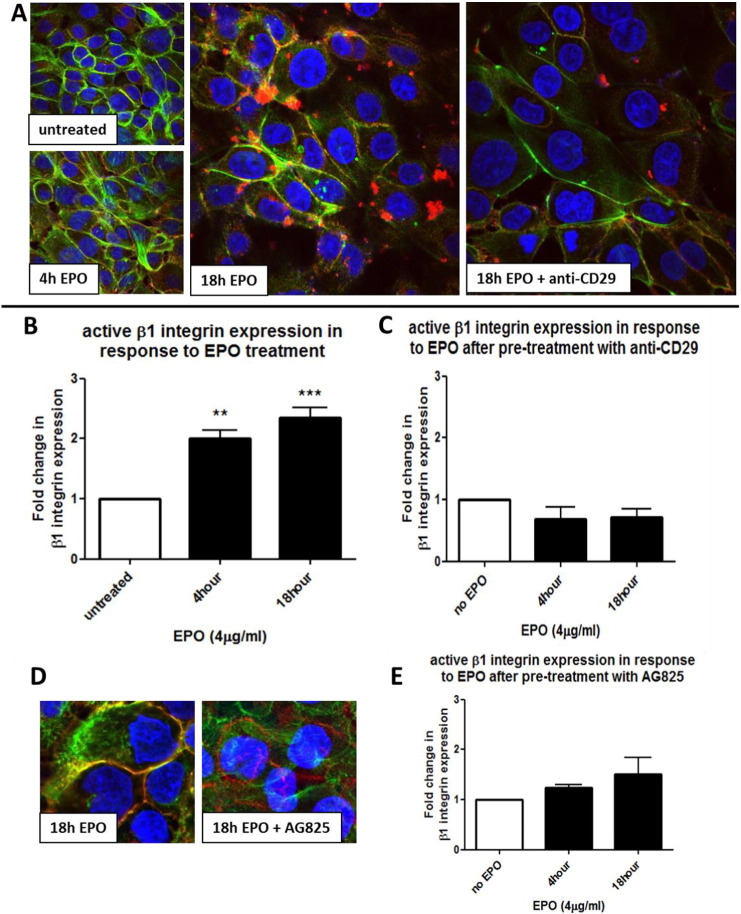
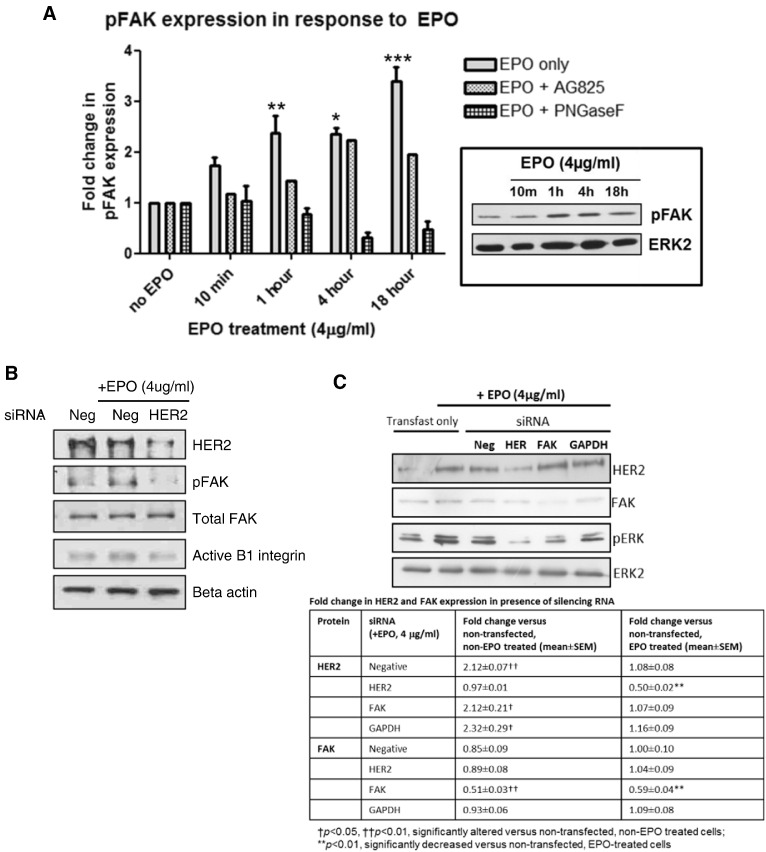
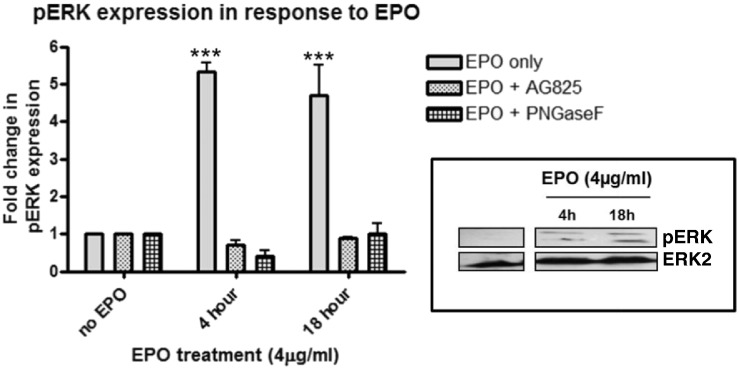
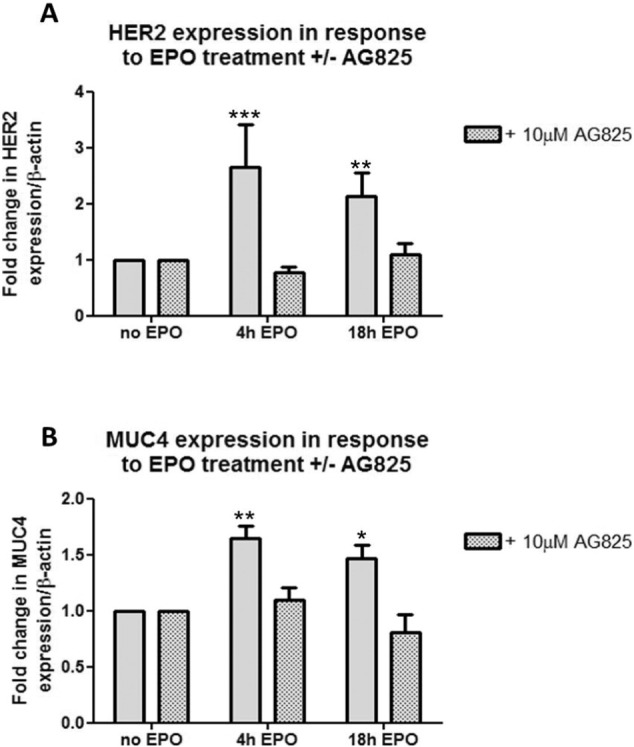
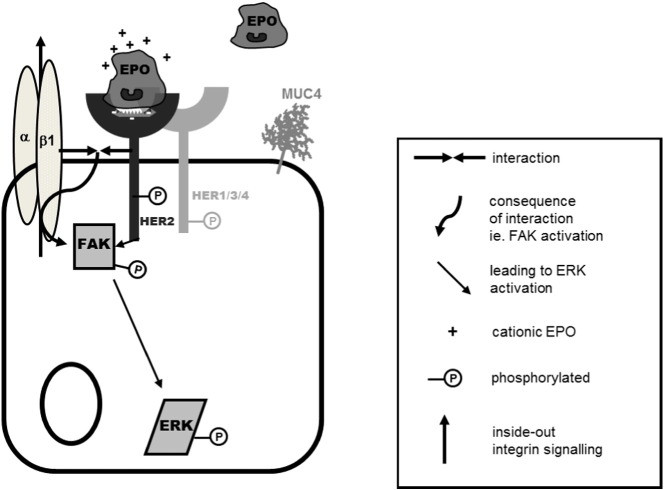
Eosinophils account for 1-3% of peripheral blood leukocytes and accumulate at sites of allergic inflammation, where they play a pathogenic role. Studies have shown that treatment with mepolizumab (an anti-IL-5 monoclonal antibody) is beneficial to patients with severe eosinophilic asthma, however, the mechanism of precisely how eosinophils mediate these pathogenic effects is uncertain. Eosinophils contain several cationic granule proteins, including Eosinophil Peroxidase (EPO). The main significance of this work is the discovery of EPO as a novel ligand for the HER2 receptor. Following HER2 activation, EPO induces activation of FAK and subsequent activation of β1-integrin, via inside-out signaling. This complex results in downstream activation of ERK1/2 and a sustained up regulation of both MUC4 and the HER2 receptor. These data identify a receptor for one of the eosinophil granule proteins and demonstrate a potential explanation of the proliferative effects of eosinophils.
Keywords: Eosinophil peroxidase; HER2; MUC4; β1-integrin.
Copyright © 2016 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Eosinophil peroxidase signals via epidermal growth factor-2 to induce cell proliferation.Am J Respir Cell Mol Biol. 2011 Nov;45(5):946-52. doi: 10.1165/rcmb.2010-0454OC. Epub 2011 Mar 31. Am J Respir Cell Mol Biol. 2011. PMID: 21454806
-
Axed MUC4 (MUC4/X) aggravates pancreatic malignant phenotype by activating integrin-β1/FAK/ERK pathway.Biochim Biophys Acta Mol Basis Dis. 2018 Aug;1864(8):2538-2549. doi: 10.1016/j.bbadis.2018.05.008. Epub 2018 May 16. Biochim Biophys Acta Mol Basis Dis. 2018. PMID: 29777904 Free PMC article.
-
Uncovering a new role for peroxidase enzymes as drivers of angiogenesis.Int J Biochem Cell Biol. 2015 Nov;68:128-38. doi: 10.1016/j.biocel.2015.09.006. Epub 2015 Sep 18. Int J Biochem Cell Biol. 2015. PMID: 26386352
-
[Research on the correlation between mechanical signaling molecules of hypertrophic scar and post injury time in burn patients in scar remodeling stage].Zhonghua Shao Shang Za Zhi. 2018 Oct 20;34(10):690-695. doi: 10.3760/cma.j.issn.1009-2587.2018.10.008. Zhonghua Shao Shang Za Zhi. 2018. PMID: 30369136 Chinese.
-
Expression and function of beta 1 integrins on human eosinophils.Mem Inst Oswaldo Cruz. 1997;92 Suppl 2:157-64. doi: 10.1590/s0074-02761997000800021. Mem Inst Oswaldo Cruz. 1997. PMID: 9698928 Review.
Cited by
-
Specific MicroRNA Pattern in Colon Tissue of Young Children with Eosinophilic Colitis.Int J Mol Sci. 2017 May 12;18(5):1050. doi: 10.3390/ijms18051050. Int J Mol Sci. 2017. PMID: 28498330 Free PMC article.
-
A new dawn for eosinophils in the tumour microenvironment.Nat Rev Cancer. 2020 Oct;20(10):594-607. doi: 10.1038/s41568-020-0283-9. Epub 2020 Jul 16. Nat Rev Cancer. 2020. PMID: 32678342 Review.
-
Human eosinophils and mast cells: Birds of a feather flock together.Immunol Rev. 2018 Mar;282(1):151-167. doi: 10.1111/imr.12638. Immunol Rev. 2018. PMID: 29431215 Free PMC article. Review.
-
Eosinophils increase airway sensory nerve density in mice and in human asthma.Sci Transl Med. 2018 Sep 5;10(457):eaar8477. doi: 10.1126/scitranslmed.aar8477. Sci Transl Med. 2018. PMID: 30185653 Free PMC article.
-
Biological Mechanisms and Therapeutic Opportunities in Mammographic Density and Breast Cancer Risk.Cancers (Basel). 2021 Oct 27;13(21):5391. doi: 10.3390/cancers13215391. Cancers (Basel). 2021. PMID: 34771552 Free PMC article. Review.
References
-
- Bousquet J., Chanez P., Lacoste J.Y., Barneon G., Ghavanian N., Enander I., Venge P., Ahlstedt S., Simony-Lafontaine J., Godard P. Eosinophilic inflammation in asthma. N. Engl. J. Med. 1990;323:1033–1039. - PubMed
-
- Kay A.B. Eosinophils and asthma. N. Engl. J. Med. 1991;324:1514–1515. - PubMed
-
- Saeed W., Badar A., Hussain M.M., Aslam M. Eosinophils and eosinophil products in asthma. J. Ayub Med. Coll. Abbottabad. 2002;14:49–55. - PubMed
-
- Costello R.W., Schofield B.H., Kephart G.M., Gleich G.J., Jacoby D.B., Fryer A.D. Localization of eosinophils to airway nerves and effect on neuronal M2 muscarinic receptor function. Am. J. Phys. 1997;273:L93–103. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
