

Biallelic PPA2 Mutations Cause Sudden Unexpected Cardiac Arrest in Infancy
- PMID: 27523598
- PMCID: PMC5010643
- DOI: 10.1016/j.ajhg.2016.06.021
Biallelic PPA2 Mutations Cause Sudden Unexpected Cardiac Arrest in Infancy
Abstract
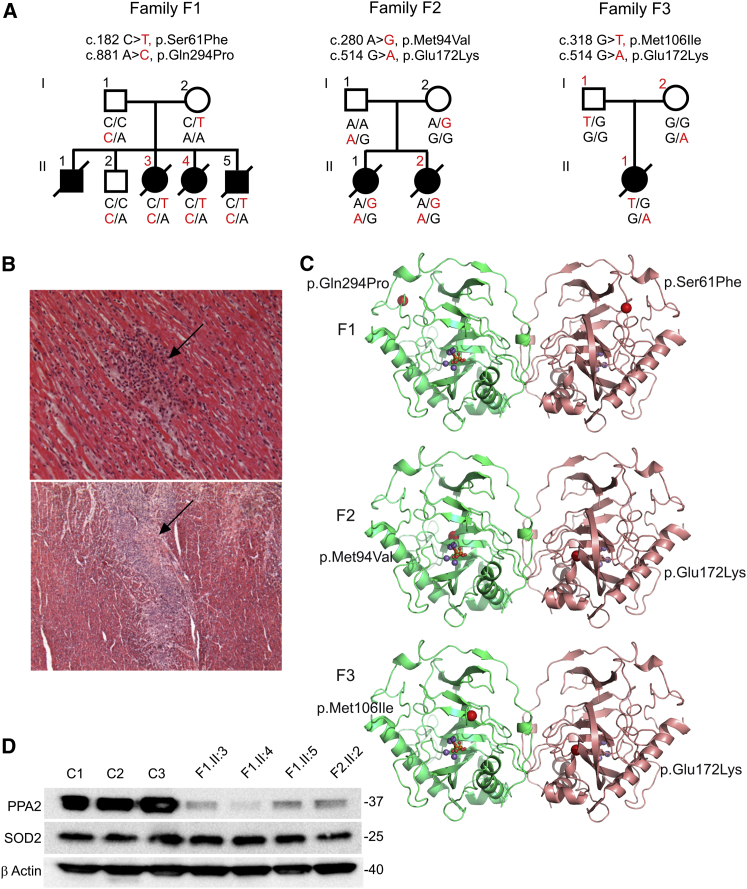
Sudden unexpected death in infancy occurs in apparently healthy infants and remains largely unexplained despite thorough investigation. The vast majority of cases are sporadic. Here we report seven individuals from three families affected by sudden and unexpected cardiac arrest between 4 and 20 months of age. Whole-exome sequencing revealed compound heterozygous missense mutations in PPA2 in affected infants of each family. PPA2 encodes the mitochondrial pyrophosphatase, which hydrolyzes inorganic pyrophosphate into two phosphates. This is an essential activity for many biosynthetic reactions and for energy metabolism of the cell. We show that deletion of the orthologous gene in yeast (ppa2Δ) compromises cell viability due to the loss of mitochondria. Expression of wild-type human PPA2, but not PPA2 containing the mutations identified in affected individuals, preserves mitochondrial function in ppa2Δ yeast. Using a regulatable (doxycycline-repressible) gene expression system, we found that the pathogenic PPA2 mutations rapidly inactivate the mitochondrial energy transducing system and prevent the maintenance of a sufficient electrical potential across the inner membrane, which explains the subsequent disappearance of mitochondria from the mutant yeast cells. Altogether these data demonstrate that PPA2 is an essential gene in yeast and that biallelic mutations in PPA2 cause a mitochondrial disease leading to sudden cardiac arrest in infants.
Copyright © 2016 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Fleming P.J., Blair P.S., Pease A. Sudden unexpected death in infancy: aetiology, pathophysiology, epidemiology and prevention in 2015. Arch. Dis. Child. 2015;100:984–988. - PubMed
-
- Hertz C.L., Christiansen S.L., Larsen M.K., Dahl M., Ferrero-Miliani L., Weeke P.E., Pedersen O., Hansen T., Grarup N., Ottesen G.L. Genetic investigations of sudden unexpected deaths in infancy using next-generation sequencing of 100 genes associated with cardiac diseases. Eur. J. Hum. Genet. 2016 - PMC - PubMed
-
- Klaver E.C., Versluijs G.M., Wilders R. Cardiac ion channel mutations in the sudden infant death syndrome. Int. J. Cardiol. 2011;152:162–170. - PubMed
-
- van Rijt W.J., Koolhaas G.D., Bekhof J., Heiner Fokkema M.R., de Koning T.J., Visser G., Schielen P.C., van Spronsen F.J., Derks T.G. Inborn errors of metabolism that cause sudden infant death: a systematic review with implications for population neonatal screening programmes. Neonatology. 2016;109:297–302. - PubMed
-
- Opdal S.H., Rognum T.O. Gene variants predisposing to SIDS: current knowledge. Forensic Sci. Med. Pathol. 2011;7:26–36. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
