

Genome-wide characteristics of de novo mutations in autism
- PMID: 27525107
- PMCID: PMC4980121
- DOI: 10.1038/npjgenmed.2016.27
Genome-wide characteristics of de novo mutations in autism
Abstract
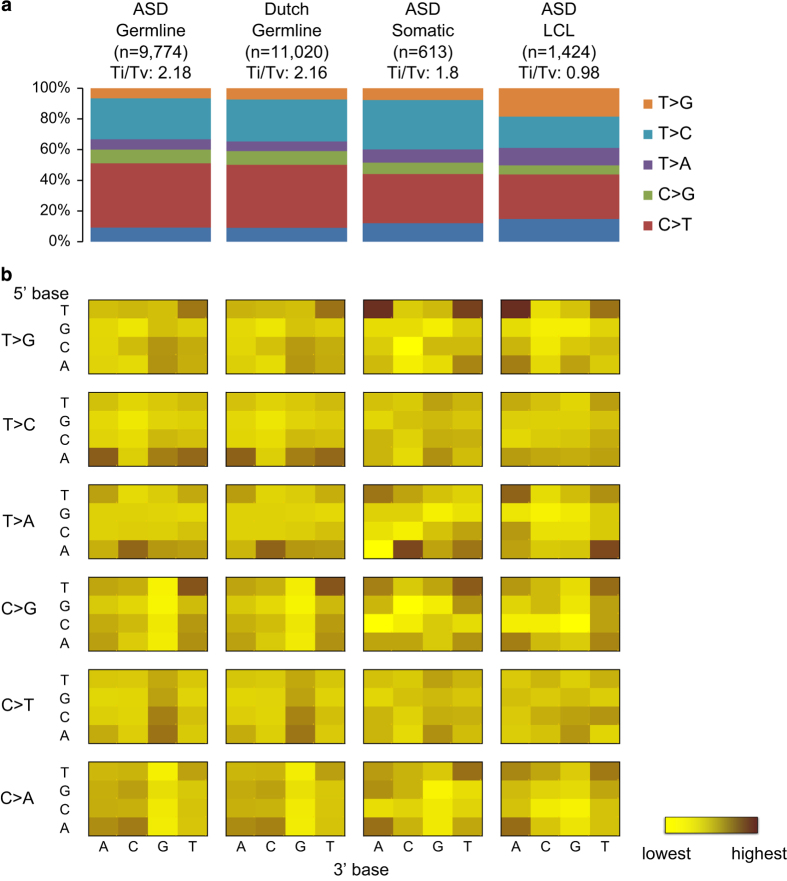
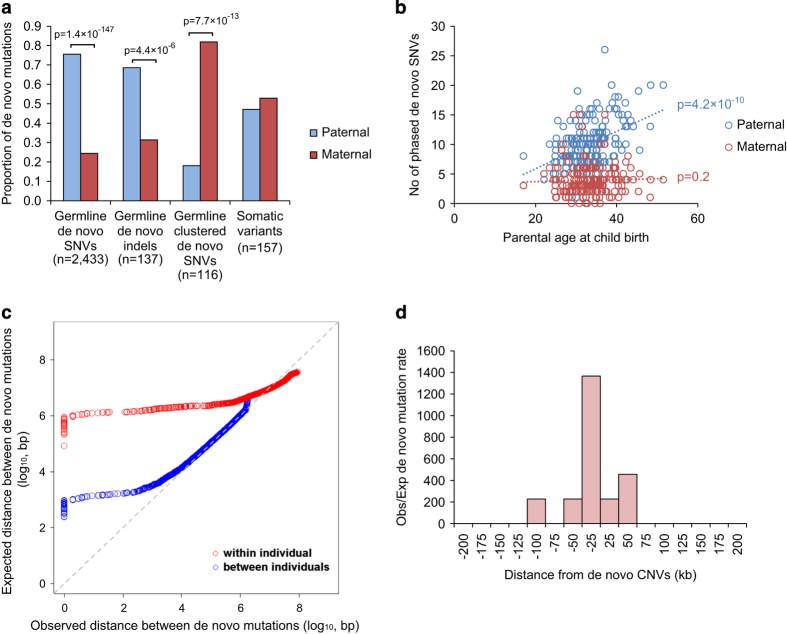
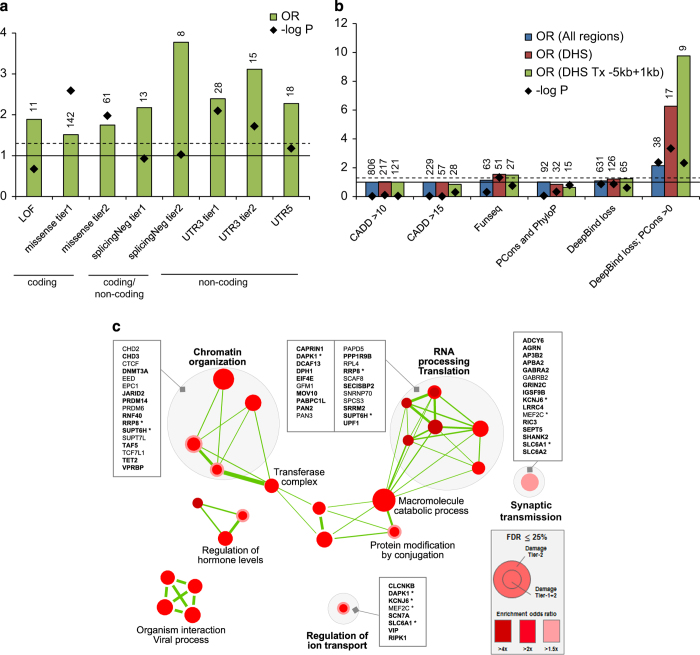
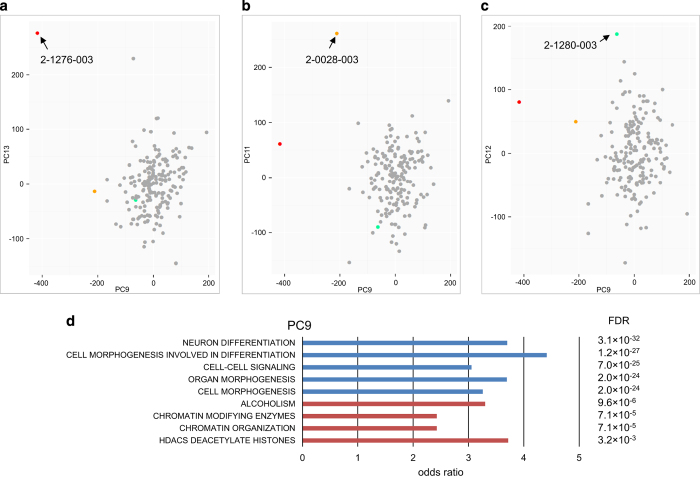
De novo mutations (DNMs) are important in Autism Spectrum Disorder (ASD), but so far analyses have mainly been on the ~1.5% of the genome encoding genes. Here, we performed whole genome sequencing (WGS) of 200 ASD parent-child trios and characterized germline and somatic DNMs. We confirmed that the majority of germline DNMs (75.6%) originated from the father, and these increased significantly with paternal age only (p=4.2×10-10). However, when clustered DNMs (those within 20kb) were found in ASD, not only did they mostly originate from the mother (p=7.7×10-13), but they could also be found adjacent to de novo copy number variations (CNVs) where the mutation rate was significantly elevated (p=2.4×10-24). By comparing DNMs detected in controls, we found a significant enrichment of predicted damaging DNMs in ASD cases (p=8.0×10-9; OR=1.84), of which 15.6% (p=4.3×10-3) and 22.5% (p=7.0×10-5) were in the non-coding or genic non-coding, respectively. The non-coding elements most enriched for DNM were untranslated regions of genes, boundaries involved in exon-skipping and DNase I hypersensitive regions. Using microarrays and a novel outlier detection test, we also found aberrant methylation profiles in 2/185 (1.1%) of ASD cases. These same individuals carried independently identified DNMs in the ASD risk- and epigenetic- genes DNMT3A and ADNP. Our data begins to characterize different genome-wide DNMs, and highlight the contribution of non-coding variants, to the etiology of ASD.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Fombonne, E. Epidemiology of pervasive developmental disorders. Pediatr. Res. 65, 591–598 (2009). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
