

Tobacco Smoke-Induced Hepatic Injury with Steatosis, Inflammation, and Impairments in Insulin and Insulin-Like Growth Factor Signaling
- PMID: 27525191
- PMCID: PMC4979551
- DOI: 10.4172/2161-0681.1000269
Tobacco Smoke-Induced Hepatic Injury with Steatosis, Inflammation, and Impairments in Insulin and Insulin-Like Growth Factor Signaling
Abstract
Background: Alcoholic liver disease (ALD) is associated with impairments in hepatic insulin and insulin-like growth factor (IGF) signaling through cell growth, survival, and metabolic pathways. Since not all heavy drinkers develop ALD, co-factors may be important. Epidemiologic data indicate that most heavy drinkers smoke tobacco and experimental data revealed that low-level nitrosamine exposures, including those from tobacco, can cause steatohepatitis with hepatic insulin/IGF resistance and exacerbate ALD. We hypothesize that cigarette smoke (CS) exposures also cause liver injury with impaired hepatic insulin/IGF signaling, and thereby contribute to ALD.
Methods: Adult male A/J mice were exposed to air for 8 weeks (A8), CS for 4 (CS4) or 8 (CS8) weeks, or CS for 8 weeks with 2 weeks recovery (CS8+R).
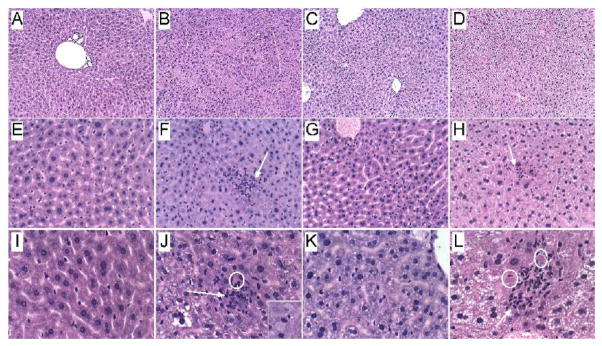
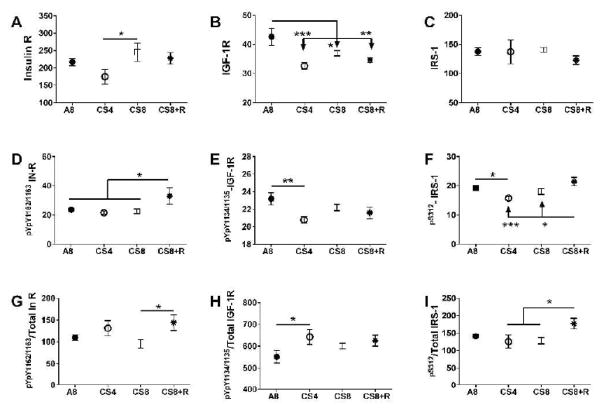
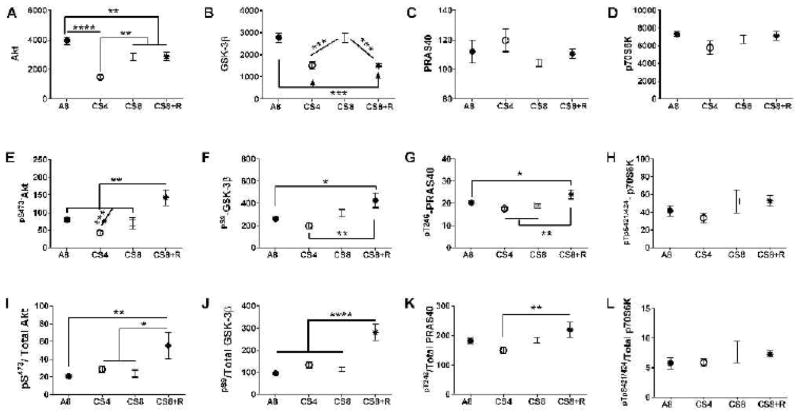
Results: CS exposures caused progressive liver injury with disruption of the normal hepatic chord architecture, lobular inflammation, apoptosis or necrosis, micro-steatosis, sinusoidal dilatation, and nuclear pleomorphism. Histopathological liver injury scores increased significantly from A8 to CS4 and then further to CS8 (P<0.0001). The mean histological grade was also higher in CS8+R relative to A8 (P<0.0001) but lower than in CS4, reflecting partial resolution of injury by CS withdrawal. CS exposures impaired insulin and IGF-1 signaling through IRS-1, Akt, GSK-3β, and PRAS40. Livers from CS8+R mice had normalized or elevated levels of insulin receptor, pYpY-Insulin-R, 312S-IRS-1, 473S-Akt, S9-GSK-3β, and pT246-PRAS40 relative to A8, CS4, or CS8, reflecting partial recovery.
Conclusion: CS-mediated liver injury and steatohepatitis with impairments in insulin/IGF signalling are reminiscent of the findings in ALD. Therefore, CS exposures (either first or second-hand) may serve as a co-factor in ALD. The persistence of several abnormalities following CS exposure cessation suggests that some aspects of CS-mediated hepatic metabolic dysfunction are not readily reversible.
Keywords: Cigarette smoke; Insulin signaling; Mouse model; Steatohepatitis; Tobacco.
Figures




References
-
- Serdula MK, Brewer RD, Gillespie C, Denny CH, Mokdad A. Trends in alcohol use and binge drinking, 1985–1999: results of a multistate survey. Am J Prev Med. 2004;26:294–298. - PubMed
-
- Centers for Disease Control and Prevention (CDC) Alcohol-attributable deaths and years of potential life lost among American Indians and Alaska Natives--United States, 2001–2005. MMWR Morb Mortal Wkly Rep. 2008;57:938–941. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous